
Graphical Abstract
Abstract
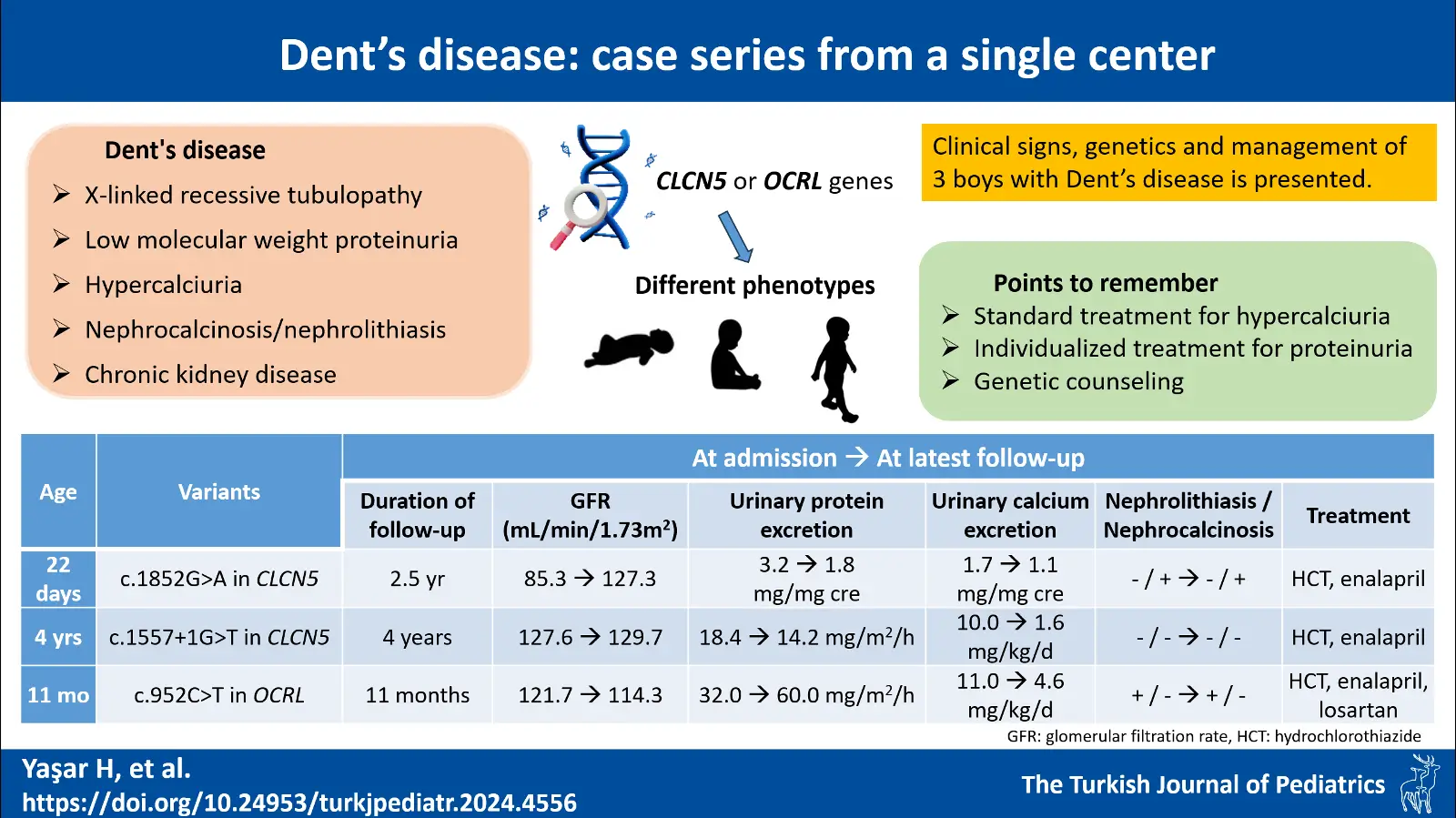
Background. Dent’s disease (DD) is a rare X-linked recessive tubulopathy characterized by low molecular weight proteinuria, hypercalciuria, nephrocalcinosis/nephrolithiasis and chronic kidney disease. With this manuscript, we reported three patients diagnosed as DD in our department in the last 10 years and thereby described the genetics, pathophysiology, clinical presentation, course and management of the disease.
Cases. The first case was a male newborn who was consulted to our department after medullary nephrocalcinosis was detected. The second case was a 4-year-old boy who was treated with a diagnosis of urinary tract infection but was found to have proteinuria. Our last case was an 11-month-old male infant who was being followed up for recurrent urinary tract infection and who had millimetric crystalloids in the renal collecting system. Proteinuria and hypercalciuria were present in all cases. Variants were observed in the CLCN5 gene for the first two cases (c.1852G>A and c.1557+1G>T, respectively) and OCRL gene (c.952C>T) for the last case. All patients were recommended oral hydration and a low-salt diet, and hydrochlorothiazide and enalapril were started. No deterioration in kidney function was observed in any patient.
Conclusion. DD is a disease that shows different phenotypes even among individuals with mutations in the same gene. Therefore, it should be considered in all patients with hypercalciuria, proteinuria, nephrolithiasis or nephrocalcinosis with/without proximal tubular dysfunction especially in the early childhood period. Classical treatments for hypercalciuria should be utilized, and a patient-based treatment plan should be drawn especially for proteinuria.
Keywords: chronic kidney disease, hypercalciuria, nephrocalcinosis, nephrolithiasis, proteinuria
References
- Blanchard A, Curis E, Guyon-Roger T, et al. Observations of a large Dent disease cohort. Kidney Int 2016; 90: 430-439. https://doi.org/10.1016/j.kint.2016.04.022
- Leggatt G, Gast C, Gilbert RD, Veighey K, Rahman T, Ennis S. Hemizygous loss of function mutations in CLCN5 causing end-stage kidney disease without Dent disease phenotype. Clin Kidney J 2022; 16: 192-194. https://doi.org/10.1093/ckj/sfac127
- Lieske JC, Milliner DS, Beara-Lasic L, Harris P, Cogal A, Abrash E. Dent disease. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. Gene Reviews. Seattle, WA: University of Washington; 2012: 1993-2023.
- Jin YY, Huang LM, Quan XF, Mao JH. Dent disease: classification, heterogeneity and diagnosis. World J Pediatr 2021; 17: 52-57. https://doi.org/10.1007/s12519-020-00357-1
- Zaniew M, Mizerska-Wasiak M, Załuska-Leśniewska I, et al. Dent disease in Poland: what we have learned so far? Int Urol Nephrol 2017; 49: 2005-2017. https://doi.org/10.1007/s11255-017-1676-x
- Dent CE, Friedman M. Hypercalcuric rickets associated with renal tubular damage. Arch Dis Child 1964; 39: 240-249. https://doi.org/10.1136/adc.39.205.240
- Zaniew M, Bökenkamp A, Kolbuc M, et al. Long-term renal outcome in children with OCRL mutations: retrospective analysis of a large international cohort. Nephrol Dial Transplant 2018; 33: 85-94. https://doi.org/10.1093/ndt/gfw350
- Gianesello L, Del Prete D, Anglani F, Calò LA. Genetics and phenotypic heterogeneity of Dent disease: the dark side of the moon. Hum Genet 2021; 140: 401-421. https://doi.org/10.1007/s00439-020-02219-2
- Fervenza FC. A patient with nephrotic-range proteinuria and focal global glomerulosclerosis. Clin J Am Soc Nephrol 2013; 8: 1979-1987. https://doi.org/10.2215/CJN.03400313
- Marzuillo P, Piccolo V, Mascolo M, et al. Patients affected by dent disease 2 could be predisposed to hidradenitis suppurativa. J Eur Acad Dermatol Venereol 2018; 32: e309-e311. https://doi.org/10.1111/jdv.14860
- Devuyst O, Christie PT, Courtoy PJ, Beauwens R, Thakker RV. Intra-renal and subcellular distribution of the human chloride channel, CLC-5, reveals a pathophysiological basis for Dent’s disease. Hum Mol Genet 1999; 8: 247-257. https://doi.org/10.1093/hmg/8.2.247
- Günther W, Piwon N, Jentsch TJ. The ClC-5 chloride channel knock-out mouse - an animal model for Dent’s disease. Pflugers Arch 2003; 445: 456-462. https://doi.org/10.1007/s00424-002-0950-6
- Piwon N, Günther W, Schwake M, Bösl MR, Jentsch TJ. ClC-5 Cl- -channel disruption impairs endocytosis in a mouse model for Dent’s disease. Nature 2000; 408: 369-373. https://doi.org/10.1038/35042597
- De Matteis MA, Staiano L, Emma F, Devuyst O. The 5-phosphatase OCRL in Lowe syndrome and Dent disease 2. Nat Rev Nephrol 2017; 13: 455-470. https://doi.org/10.1038/nrneph.2017.83
- Vicinanza M, Di Campli A, Polishchuk E, et al. OCRL controls trafficking through early endosomes via PtdIns4,5P2-dependent regulation of endosomal actin. EMBO J 2011; 30: 4970-4985. https://doi.org/10.1038/emboj.2011.354
- Wu G, Zhang W, Na T, Jing H, Wu H, Peng JB. Suppression of intestinal calcium entry channel TRPV6 by OCRL, a lipid phosphatase associated with Lowe syndrome and Dent disease. Am J Physiol Cell Physiol 2012; 302: C1479-C1491. https://doi.org/10.1152/ajpcell.00277.2011
- Gianesello L, Arroyo J, Del Prete D, et al. Genotype phenotype correlation in Dent disease 2 and review of the literature: OCRL gene pleiotropism or extreme phenotypic variability of lowe syndrome? Genes (Basel) 2021; 12: 1597. https://doi.org/10.3390/genes12101597
- National Library of Medicine. ClinVar-Genomic variation as it relates to human health. Available at: https://www.ncbi.nlm.nih.gov/clinvar/variation/758735/ (Accessed on August 10, 2024).
- gnomAD browser. SNVX-50090223-G-A(GRCh38), in silico predictors. Available at: https://gnomad.broadinstitute.org/variant/X-50090223-G-A?dataset=gnomad_r4 (Accessed on September 28, 2024).
- Katsonis P, Wilhelm K, Williams A, Lichtarge O. Genome interpretation using in silico predictors of variant impact. Hum Genet 2022; 141: 1549-1577. https://doi.org/10.1007/s00439-022-02457-6
- Tosetto E, Ceol M, Mezzabotta F, et al. Novel mutations of the CLCN5 gene including a complex allele and A 5’ UTR mutation in Dent disease 1. Clin Genet 2009; 76: 413-416. https://doi.org/10.1111/j.1399-0004.2009.01212.x
- Sekine T, Komoda F, Miura K, et al. Japanese Dent disease has a wider clinical spectrum than Dent disease in Europe/USA: genetic and clinical studies of 86 unrelated patients with low-molecular-weight proteinuria. Nephrol Dial Transplant 2014; 29: 376-384. https://doi.org/10.1093/ndt/gft394
- Ehlayel AM, Copelovitch L. Update on Dent disease. Pediatr Clin North Am 2019; 66: 169-178. https://doi.org/10.1016/j.pcl.2018.09.003
- van Berkel Y, Ludwig M, van Wijk JAE, Bökenkamp A. Proteinuria in Dent disease: a review of the literature. Pediatr Nephrol 2017; 32: 1851-1859. https://doi.org/10.1007/s00467-016-3499-x
- Vaisbich MH, Henriques L dos S, Igarashi T, Sekine T, Seki G, Koch VH. The long-term use of enalapril and hydrochlorothiazide in two novel mutations patients with Dent’s disease type 1. J Bras Nefrol 2012; 34: 78-81.
- Solanki AK, Arif E, Morinelli T, et al. A Novel CLCN5 mutation associated with focal segmental glomerulosclerosis and podocyte injury. Kidney Int Rep 2018; 3: 1443-1453. https://doi.org/10.1016/j.ekir.2018.06.003
- Deng H, Zhang Y, Xiao H, et al. Phenotypic spectrum and antialbuminuric response to angiotensin converting enzyme inhibitor and angiotensin receptor blocker therapy in pediatric Dent disease. Mol Genet Genomic Med 2020; 8: e1306. https://doi.org/10.1002/mgg3.1306
- Borghi L, Nouvenne A, Meschi T. Nephrolithiasis and urinary tract infections: ‘the chicken or the egg’ dilemma? Nephrol Dial Transplant 2012; 27: 3982-3984. https://doi.org/10.1093/ndt/gfs395
- Cetin N, Gencler A, Kavaz Tufan A. Risk factors for development of urinary tract infection in children with nephrolithiasis. J Paediatr Child Health 2020; 56: 76-80. https://doi.org/10.1111/jpc.14495
Copyright and license
Copyright © 2024 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









