
Graphical Abstract

Abstract
Background. Infantile fibrosarcoma is a rapidly growing soft tissue tumor, often managed by surgical resection, with chemotherapy and radiotherapy as additional options. Due to the high local aggressiveness and surgical morbidity, targeted therapies like larotrectinib can enhance quality of life and preserve organs, particularly in limb-threatening cases. Here, we present three cases where larotrectinib prevented mutilating surgeries.
Cases. The first patient presented antenatally with a 75 x 48 mm oral floor mass, severely narrowing the airway. Surgery was unfeasible due to invasion of vital organs, and complications arose with conventional chemotherapy. Following detection of an ETV6-NTRK3 fusion, larotrectinib was initiated, resulting in complete regression after two years. The second patient had a 42 x 37 mm right-hand tumor confirmed as infantile fibrosarcoma, for which initial treatment suggested amputation. After identifying an ETV6-NTRK3 fusion and failing to respond to chemotherapy, larotrectinib led to significant regression by year two, preserving hand function. The third patient presented with a 56 x 55 mm right foot mass at birth. Chemotherapy proved ineffective, and larotrectinib was initiated due to an ETV6-NTRK fusion signal, ultimately achieving near total regression within one year and avoiding amputation. All three cases demonstrated successful outcomes with targeted therapy.
Conclusions. These cases emphasize the importance of advanced molecular studies, like next-generation sequencing, for childhood tumors and integrating research with clinical trials. tropomyosin receptor kinase inhibitor larotrectinib may offer a safe and effective alternative to chemotherapy for NTRK fusion-positive, metastatic, or unresectable tumors.
Keywords: infantile fibrosarcoma, larotrectinib, TRK inhibitors
Introduction
Fibrosarcoma, a rare malignant soft-tissue tumor, is classified into infantile (congenital) and adult types. Infantile fibrosarcoma (IFS) typically presents in children under 1 year, most often in the extremities but also in the head, neck, or abdomen.1 The five-year survival rate for localized IFS is 89%, with an event-free survival rate of 81%.2 Treatment aims to achieve a cure with minimal long-term side effects.
The primary treatment for IFS is conservative surgical resection. When direct surgery is not feasible, systemic treatments, such as chemotherapy, are used to reduce tumor size and facilitate secondary resection. Standard chemotherapies like vincristine and actinomycin-D are commonly used in Europe; however, they have limitations, highlighting the need for safer alternatives. Aggressive therapies, including alkylating agents, anthracyclines, mutilating surgeries, and radiotherapy, are generally avoided due to severe long-term effects and are reserved for relapse or recurrence.3,4
The ETS Variant Transcription Factor 6- Neurotrophic Receptor Tyrosine Kinase 3 (ETV6-NTRK3) fusion is a major oncogenic driver in up to 90% of IFS cases.5 Initially used for diagnostic purposes, it has become a valuable therapeutic target. Selective tropomyosin receptor kinase (TRK) inhibitors are particularly promising for pediatric patients with neurotrophic tyrosine receptor kinase (NTRK) fusion-positive tumors, where chemotherapy and radiotherapy are less ideal. These inhibitors are recommended when tumors are metastatic, resection would result in significant morbidity, or alternative treatments are unsatisfactory or have failed. TRK inhibitors are now increasingly favored as the primary treatment for most IFS patients.6
Randomized controlled trials are challenging for rare conditions like IFS, limiting robust evidence. Consequently, recommendations for the TRK inhibitor larotrectinib rely on cumulative case reports.4,7-12 Even a small case series, such as three patients, can offer meaningful insights and enrich the literature. We report three cases showing that larotrectinib may be a valid therapeutic option for newborn and infant patients with IFS.
Case Reports
Case 1
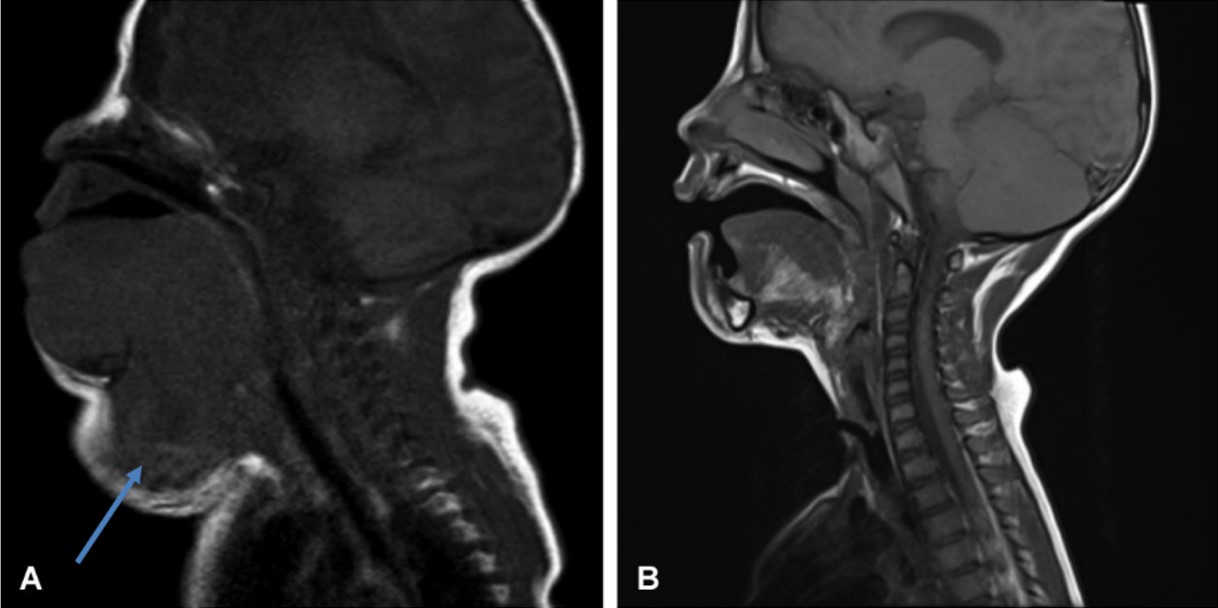
The first patient was diagnosed antenatally with a suspicious neck mass extending from the tongue root. Fetal magnetic resonance imaging (MRI) suggested a sarcomatous tumor (Fig. 1A), and postnatal tru-cut biopsy confirmed a spindle-cell mesenchymal tumor. An ETV6-NTRK3 fusion was detected in 30% of tumor cells, leading to a diagnosis of IFS.
Primary surgery was not feasible due to vital organ invasion. Vincristine and actinomycin D treatment began immediately, but severe airway narrowing at 18 days required an open tracheostomy. After the third chemotherapy cycle, the regimen was changed to vincristine and cyclophosphamide due to veno-occlusive disease (VOD) and slow response, but treatment was discontinued soon because of toxicity.
Larotrectinib was initiated as a single agent three months after diagnosis, following the identification of the ETV6-NTRK3 fusion. The patient’s clinical condition significantly improved, with the best response noted by week 8 of larotrectinib.
Larotrectinib treatment was continued for two years. After stopping the treatment, the patient was evaluated for closure of the tracheostomy and could be fed orally. The patient has since been followed up for one year and has shown a complete response (Fig. 1B and Table I).
| Chemo: chemotherapy, VOD: veno-occlusive disease, CR: complete remission, NED: no evidence of disease. | |||
| Table I. Clinical and radiological features of three cases. | |||
| Features | Case 1 | Case 2 | Case 3 |
| Age at diagnosis | 2 days old | 20 days old | First day of birth |
| Sex | Male | Female | Female |
| Tumor localization |
Floor of mouth and tongue muscle Oropharynx Deep lobe of left parotid gland to hypopharynx Narrowing in airway at these levels |
Surrounding second metacarpi on the hand Infiltrating adductor pollisus muscle group, intraosseous muscles Ending with distal row of carpal bones |
Starting from tarsal bone level in right foot Extending distally, surrounding tarsal and metatarsal bones 360 degrees |
| Size | 75 x 48 mm | 42 x 37 mm | 56 x 55 mm |
| Previous treatment |
Vincristine Cyclophosphamide Actinomycin D |
Vincristine Cyclophosphamide Actinomycin D |
Vincristine Cyclophosphamide Actinomycin D |
| Initiation of larotrectinib |
Third month of chemo | After 3 cycles of chemo | After 2 cycles of chemo |
| Larotrectinib dosage | 100 mg/m2 twice daily | 100 mg/m2 twice daily | 100 mg/m2 twice daily |
| Chemo complications | VOD | None | None |
| Reason for chemo discontinuation |
VOD and Slow response |
Inadequate response | Inadequate response |
| Larotrectinib related side effects | None | None | None |
| Outcome | CR | CR | Near CR |
| Duration of larotrectinib treatment |
2 years | 2 years | 1 year |
| Follow-up | NED | NED | NED |
Case 2
The second patient was a 20-day-old female admitted with a large mass in her right hand. Ultrasound identified a mass with vascular structures, and MRI revealed a 42 x 37 mm lesion surrounding the second metacarpal and infiltrating the adductor pollicis muscle group and distal carpal bones (Fig. 2A).
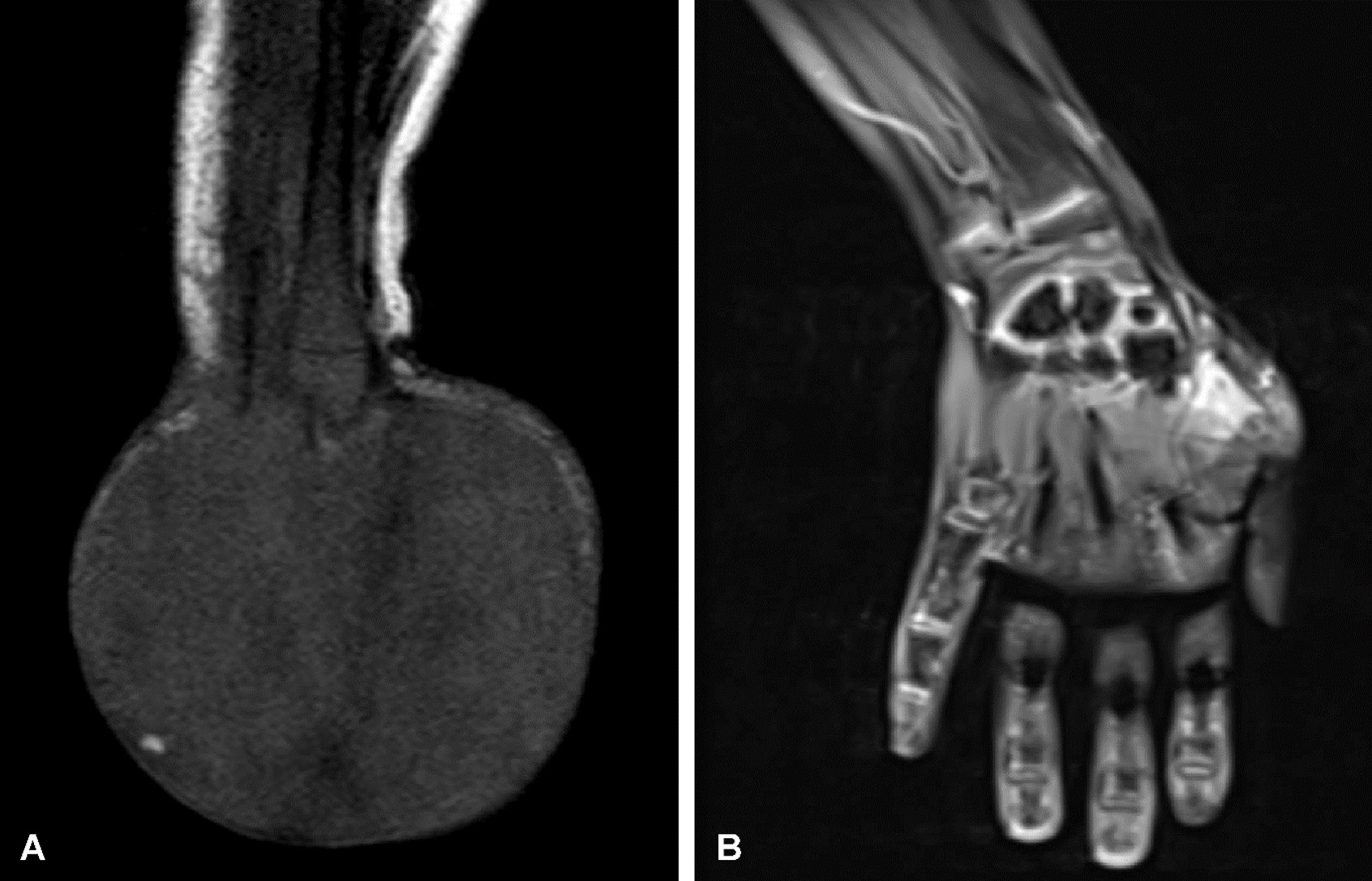
Diagnosis of IFS was confirmed by biopsy, revealing an ETV6-NTRK3 fusion in 100% of examined cells. The patient began treatment with vincristine and actinomycin D. Follow-up MRI after three chemotherapy cycles showed residual tumor without mass regression. Surgical evaluation suggested potential limb amputation; however, larotrectinib was initiated, successfully preserving the limb (Fig. 2B). One month after the initiation of treatment, a noticeable improvement in response was observed. With 2 years of larotrectinib treatment, the residual lesion completely disappeared. The patient is now 3 years old, and even at 1 year following discontinuation of treatment, she can use her extremity functionally, and there are no signs of the tumor (Table I).
Case 3
On the first postnatal day, the third female patient was treated for a mass with necrotic areas on the dorsum and sole of the right foot. Computed tomography angiography indicated a heterogeneous hypervascular lesion surrounding the tarsal and metatarsal bones, consistent with a malignant mesenchymal tumor.
MRI revealed a 56x55 mm mass originating at the tarsal bone in the right foot, surrounding the tarsal and metatarsal bones by 360 degrees (Fig. 3A). Tru-cut biopsy indicated spindle cell sarcoma, and an ETV6-NTRK fusion confirmed the diagnosis of IFS.
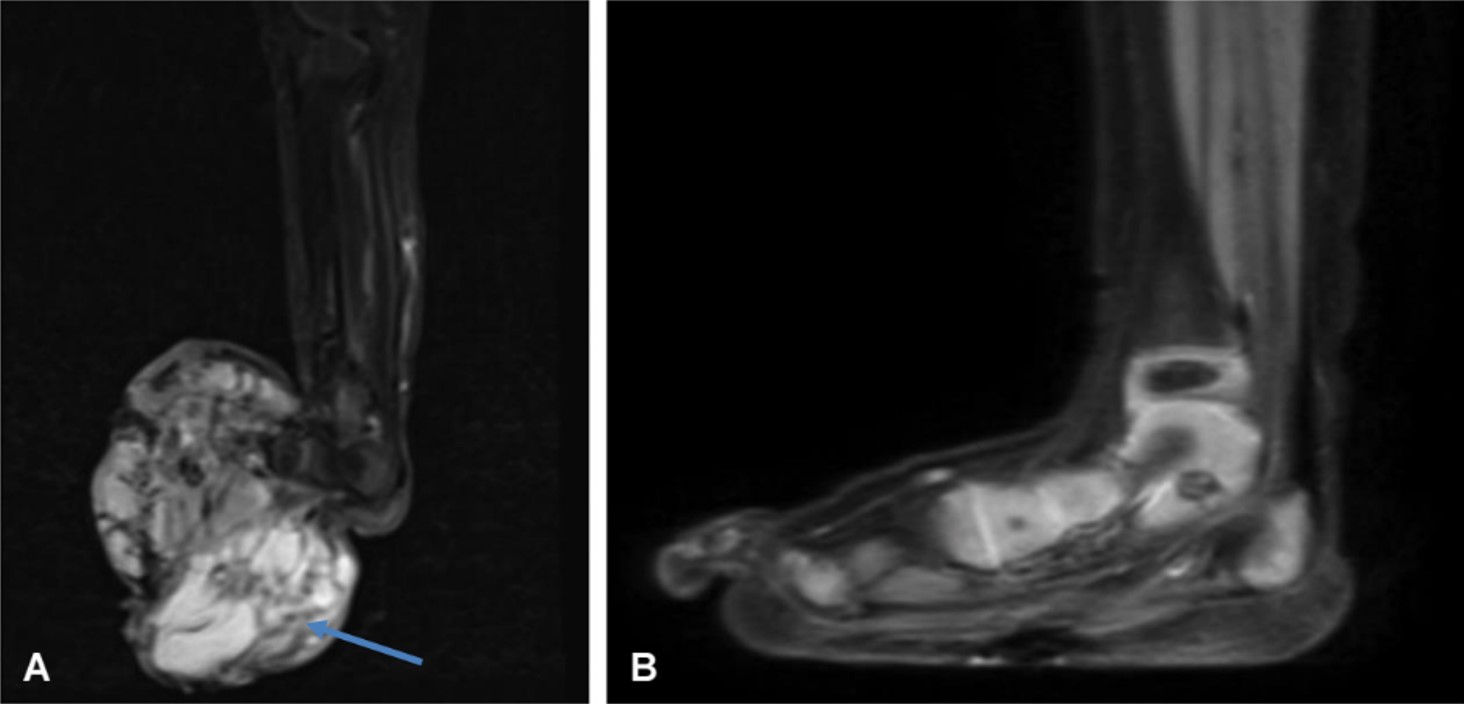
The treatment regimen initially included vincristine, actinomycin D, and cyclophosphamide; however, the tumor did not regress and was deemed unsuitable for surgery. After two cycles, the treatment was switched to larotrectinib. A reduction in macroscopic mass dimensions was noted by the second month, with the best response achieved by month three. Larotrectinib was used for one year and spared the patient from mutilating surgery (Fig. 3B and Table I). The patient no longer required surgical intervention. The larotrectinib was discontinued when nearly total regression was detected. The patient has been followed up for 2 years without treatment and can successfully use her extremity.
All families provided written informed consent for the publication.
Discussion
The results presented showed rapid decreases in tumor size during the first 3 months of treatment and the achievement of tumor remission with larotrectinib in three infants with IFS. Surgery was the primary approach in the 1980s for IFS and is still the mainstay of therapy, but it is now treated with a more multidisciplinary approach, and surgery is applied as the final step.13-15 Chemotherapy can be used as the first step, and curative effects have been observed with vincristine, actinomycin D, cyclophosphamide or ifosfamide, and etoposide.2
In 2003, Russell et al.3 conducted a literature review on IFS and identified 22 cases treated with chemotherapy either preoperatively or as the sole treatment. Among the chemotherapy agents mentioned in the studies, vincristine, cyclophosphamide, actinomycin D, and doxorubicin were the most commonly used. Additionally, six infants were treated with chemotherapy protocols that included ifosfamide and etoposide.3,16
Loh et al.17 also treated patients with similar chemotherapy. The regimens included vincristine, adriamycin, and cyclophosphamide (adria-VAC); vincristine, actinomycin-D, and cyclophosphamide (actino-VAC); and etoposide and ifosfamide. They described 11 infants who were treated for IFS over a 16-year period at the Dana-Farber Cancer Institute and Children’s Hospital, Boston. All of these regimens were tolerated well. A recent multi-institutional European study concluded that conservative surgery is the mainstay of treatment, and alkylating agent and anthracycline-free first-line chemotherapy should be used to treat unresectable tumors.2,18
Larotrectinib is a selective TRK inhibitor that can be given orally in cases of ETV6-NTRK3 fusion. In our cases, we used larotrectinib due to unresponsiveness or discontinuation of chemotherapy (one of our cases developed VOD), and two of them showed no response to chemotherapy). All of our cases were positive for ETV6-NTRK3 fusion and showed complete responses to larotrectinib. Additionally, two of our patients were saved from amputation by larotrectinib. In the other case with head and neck localization, airway patency was provided with tracheostomy, and effective results were obtained with larotrectinib treatment without the need for mutilating surgery.
The EPI VITRAKVI study was a retrospective, observational investigation (NCT05236257) that utilized historical data to evaluate the effectiveness of larotrectinib in comparison to chemotherapy-based treatments.4 Among the patients treated with larotrectinib, 49% experienced a complete response, 41% had a partial response, and 9.8% had stable disease. None of the patients discontinued treatment permanently due to drug-related side effects. Patients with IFS demonstrated significant and durable responses to larotrectinib.
The findings indicated that larotrectinib offers a therapeutic advantage over the conventional chemotherapy treatments for pediatric patients with locally advanced or metastatic IFS.4 The study revealed that larotrectinib notably extended the time until treatment failure (defined as the need for subsequent systemic therapy, radiotherapy, surgery, or death, whichever occurred first) when compared to external historical controls who were only treated with chemotherapy. The results indicate that treatment with larotrectinib reduced morbidity and the need for aggressive local therapies compared to conventional chemotherapy in children with IFS.4
Consequently, this treatment method is particularly promising for cases that are treatment resistant and ineligible for surgery. Clinical trials have already demonstrated the pantumour efficacy of larotrectinib with a favorable safety profile in patients as young as 1 month of age.19-22
The drug was administered orally on a continuous 28-day schedule to pediatric patients, primarily at a dose of 100 mg/m² (up to 100 mg) twice daily. Treatment continued until a complete response was achieved. Common adverse events reported in the literature include elevated alanine aminotransferase, anemia, and neutropenia, with no treatment-related deaths noted. We observed no drug-related side effects or compliance issues in our patients. It is necessary to consider that TRKs are involved in neural development, and the long-term neurologic effects of larotrectinib have not been clearly shown.23
Locally advanced TRK fusion sarcomas have significant morbidity with surgical resection, and larotrectinib continues to be evaluated as a presurgical therapy for children who are newly diagnosed.24,25 Larotrectinib treatment should be attempted for patients who are diagnosed with IFS and have ETV6-NTRK3 fusion, especially if the response to chemotherapy is insufficient. Nevertheless, multi-center studies are needed, and the duration of treatment should be decided according to radiological and clinical findings. Although there are case reports of limb salvage for patients with IFS in the literature, there are no robust data on the duration of treatment or the risk of recurrence after discontinuation of larotrectinib.25
Orbach et al.26 developed a consensus strategy with the Children’s Oncology Group (COG) after analyzing retrospective data for all reported patients with IFS from the European Paediatric Soft Tissue Sarcoma Study Group and Cooperative Weichteil Sarkomstudien Gruppe (CWS). The median duration of conventional chemotherapy in patients with localized disease has been reported to be 4–6 months. For patients who are initially unresectable, the optimal duration of larotrectinib administration has been defined as 4 months to 1 year, and drug discontinuation is recommended if total surgical resection is feasible.26 It is also recommended that the duration of larotrectinib use be extended for metastatic patients or patients with localized tumors that are still not suitable for resection despite tumor regression.26 It is not yet known how long larotrectinib therapy should be continued in cases of complete clinical and radiological response of the tumor without surgery.26-28
The COG ADVL1823 trial administered larotrectinib continuously in 28-day cycles until the disease progressed, the tumor became surgically resectable following a minimum of 6 cycles of therapy, or the completion of 12 cycles (for those with complete remission) or 26 cycles (for those with partial remission) of therapy.20,29 In all three of our cases, a significant response to larotrectinib was observed within the first three months. The treatment duration can vary between clinical trials. The results of attempts to discontinue therapy in these situations are not yet available.
In conclusion, our case series has presented the successful treatment of patients with IFS in different locations using a targeted agent without the need for mutilating surgery. We continued larotrectinib until complete clinical and radiological response of the tumor occurred without surgery. The medication also has a safe side effect profile. Thus, larotrectinib could emerge as an efficacious and safe alternative to chemotherapy for NTRK fusion-positive and metastatic or unresectable tumors.
Ethical approval
Informed consent was obtained from the parents or legal guardians of the patients for the publication.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Davis DD, Shah SJ, Kane SM. Fibrosarcoma. In: StatPearls. Treasure Island (FL): StatPearls Publishing, 2022. Available at: https://www.ncbi.nlm.nih.gov/books/NBK560759/
- Orbach D, Rey A, Cecchetto G, et al. Infantile fibrosarcoma: management based on the European experience. J Clin Oncol 2010; 28: 318-323. https://doi.org/10.1200/JCO.2009.21.9972
- Russell H, Hicks MJ, Bertuch AA, Chintagumpala M. Infantile fibrosarcoma: clinical and histologic responses to cytotoxic chemotherapy. Pediatr Blood Cancer 2009; 53: 23-27. https://doi.org/10.1002/pbc.21981
- Orbach D, Carton M, Khadir SK, et al. Therapeutic benefit of larotrectinib over the historical standard of care in patients with locally advanced or metastatic infantile fibrosarcoma (EPI VITRAKVI study). ESMO Open 2024; 9: 103006. https://doi.org/10.1016/j.esmoop.2024.103006
- Sandberg AA, Bridge JA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: congenital (infantile) fibrosarcoma and mesoblastic nephroma. Cancer Genet Cytogenet 2002; 132: 1-13. https://doi.org/10.1016/s0165-4608(01)00528-3
- Knezevich SR, Garnett MJ, Pysher TJ, Beckwith JB, Grundy PE, Sorensen PH. ETV6-NTRK3 gene fusions and trisomy 11 establish a histogenetic link between mesoblastic nephroma and congenital fibrosarcoma. Cancer Res 1998; 58: 5046-5048.
- Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol 2018; 19: 705-714. https://doi.org/10.1016/S1470-2045(18)30119-0
- Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med 2018; 378: 731-739. https://doi.org/10.1056/NEJMoa1714448
- Hong DS, DuBois SG, Kummar S, et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol 2020; 21: 531-540. https://doi.org/10.1016/S1470-2045(19)30856-3
- Chu P, Batson S, Hodgson M, Mitchell CR, Steenrod A. Systematic review of neurotrophic tropomyosin-related kinase inhibition as a tumor-agnostic management strategy. Future Oncol 2020; 16: 61-74. https://doi.org/10.2217/fon-2019-0534
- Lengliné E, Peron J, Vanier A, et al. Basket clinical trial design for targeted therapies for cancer: a French National Authority for Health statement for health technology assessment. Lancet Oncol 2021; 22: e430-e434. https://doi.org/10.1016/S1470-2045(21)00337-5
- Vanier A, Fernandez J, Kelley S, et al. Rapid access to innovative medicinal products while ensuring relevant health technology assessment. Position of the French National Authority for Health. BMJ Evid Based Med 2024; 29: 1-5. https://doi.org/10.1136/bmjebm-2022-112091
- Augsburger D, Nelson PJ, Kalinski T, et al. Current diagnostics and treatment of fibrosarcoma -perspectives for future therapeutic targets and strategies. Oncotarget 2017; 8: 104638-104653. https://doi.org/10.18632/oncotarget.20136
- Ferrari A, Orbach D, Sultan I, Casanova M, Bisogno G. Neonatal soft tissue sarcomas. Semin Fetal Neonatal Med 2012; 17: 231-238. https://doi.org/10.1016/j.siny.2012.05.003
- Ferrari A, Brennan B, Casanova M, et al. Pediatric non-rhabdomyosarcoma soft tissue sarcomas: standard of care and treatment recommendations from the European Paediatric Soft Tissue Sarcoma Study Group (EpSSG). Cancer Manag Res 2022; 14: 2885-2902. https://doi.org/10.2147/CMAR.S368381
- Ramphal R, Manson D, Viero S, Zielenska M, Gerstle T, Pappo A. Retroperitoneal infantile fibrosarcoma: clinical, molecular, and therapeutic aspects of an unusual tumor. Pediatr Hematol Oncol 2003; 20: 635-642.
- Loh ML, Ahn P, Perez-Atayde AR, Gebhardt MC, Shamberger RC, Grier HE. Treatment of infantile fibrosarcoma with chemotherapy and surgery: results from the Dana-Farber Cancer Institute and Children’s Hospital, Boston. J Pediatr Hematol Oncol 2002; 24: 722-726. https://doi.org/10.1097/00043426-200212000-00008
- Parida L, Fernandez-Pineda I, Uffman JK, et al. Clinical management of infantile fibrosarcoma: a retrospective single-institution review. Pediatr Surg Int 2013; 29: 703-708. https://doi.org/10.1007/s00383-013-3326-4
- Looney AM, Nawaz K, Webster RM. Tumour-agnostic therapies. Nat Rev Drug Discov 2020; 19: 383-384. https://doi.org/10.1038/d41573-020-00015-1
- Mascarenhas L, Tilburg CM, Doz F, et al. Efficacy and safety of larotrectinib in pediatric patients with tropomyosin receptor kinase (TRK) fusion-positive cancer: an expanded dataset. J Clin Oncol 2022; 40(Suppl. 16): 10030. https://doi.org/10.1200/JCO.2022.40.16_suppl.10030
- U.S. Food and Drug Administration (FDA). VITRAKVI-Accelerated Approval (COR-NDAACTION-04). 2018. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/211710s000lbl.pdf
- Chae YJ, Song YK, Chae SH, et al. Development and validation of an LC-MS/MS method for monitoring larotrectinib, a tropomyosin-related kinase inhibitor, in mouse and human plasma and application to pharmacokinetic studies. J Anal Sci Technol 2020; 11: 20. https://doi.org/10.1186/s40543-020-00219-5
- DuBois SG, Laetsch TW, Federman N, et al. The use of neoadjuvant larotrectinib in the management of children with locally advanced TRK fusion sarcomas. Cancer 2018; 124: 4241-4247. https://doi.org/10.1002/cncr.31701
- Caldwell KJ, De La Cuesta E, Morin C, Pappo A, Helmig S. A newborn with a large NTRK fusion positive infantile fibrosarcoma successfully treated with larotrectinib. Pediatr Blood Cancer 2020; 67: e28330. https://doi.org/10.1002/pbc.28330
- Lapeña LM, Caldas MCS, Ramírez C, et al. Larotrectinib as an effective therapy in congenital infantile fibrosarcoma: report of two cases. European J Pediatr Surg Rep 2022; 10: e76-e79. https://doi.org/10.1055/s-0042-1748866
- Orbach D, Sparber-Sauer M, Laetsch TW, et al. Spotlight on the treatment of infantile fibrosarcoma in the era of neurotrophic tropomyosin receptor kinase inhibitors: International consensus and remaining controversies. Eur J Cancer 2020; 137: 183-192. https://doi.org/10.1016/j.ejca.2020.06.028
- Carton M, Del Castillo JP, Colin JB, et al. Larotrectinib versus historical standard of care in patients with infantile fibrosarcoma: protocol of EPI-VITRAKVI. Future Oncol 2023; 19: 1645-1653. https://doi.org/10.2217/fon-2023-0114
- Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol 2018; 15: 731-747. https://doi.org/10.1038/s41571-018-0113-0
- Laetsch TW, Ludwig K, Hall D, et al. Phase 2 study of larotrectinib in children with newly diagnosed infantile fibrosarcoma (IFS): Children’s Oncology Group (COG) ADVL1823 cohort A. J Clin Oncol 2023; 41(Suppl. 16): 10008. https://doi.org/10.1200/JCO.2023.41.16_suppl.10008
Copyright and license
Copyright © 2025 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









