
Graphical Abstract
Abstract
Background. In transfusion-dependent forms of β-thalassemia, chronic anemia and iron overload lead to the development of oxidative stress-related DNA damage. In β-thalassemia minor (β-Tm), oxidative stress resulting from an unbalanced globin chain ratio has been documented, even in the absence of anemia and its complications. However, the status of oxidative stress-related DNA damage has not yet been elucidated. The aim of this study was to assess DNA damage in β-Tm in a pediatric population.
Material and Methods. We compared 142 children with β-Tm to 113 healthy controls, including siblings of the β-Tm individuals. The comet assay was used to assess DNA damage in peripheral blood lymphocytes. Additionally, oxidative stress markers and biochemical parameters were measured.
Results. No significant differences were observed between the β-Tm group and controls in terms of demographics, biochemical parameters, or baseline oxidative stress levels (p>0.05). In the comet assay, there was no difference in tail intensity (TI) between subjects and controls, nor between siblings with and without β-Tm (p=0.551 and p=0.655, respectively). However, when the β-Tm group was divided by age, a gradual increase in DNA damage, as measured by TI, was observed. This increase was more pronounced in the β-Tm group compared to controls.
Conclusion. We observed no significant differences in DNA damage between β-Tm individuals and controls. However, TI increased at a faster rate with age in carriers compared to non-carriers, suggesting that environmental factors might exert a more pronounced influence on the genetic integrity of individuals with a β-Tm background. Although β-Tm itself does not seem to pose a substantial genotoxic risk in childhood, our findings underscore the importance of further research into the interplay between β-Tm and other risk factors throughout life. We advocate for long-term monitoring of β-Tm children to assess the health and potential genetic consequences.
Keywords: DNA damage, oxidative stress, comet assay, children, β-thalassemia minor
Introduction
Thalassemias, resulting from inherited defects in the production of hemoglobin, are among the most common genetic disorders worldwide.1-4 They are characterized by absent or reduced synthesis of one or more globin chains. Degradation of unpaired chains in erythrocytes causes premature cell death. Thalassemias are designated depending on the affected globin chain. In β-thalassemia, β-globin chain production is impaired due to mutations in one or both of the β-globin genes, each located on a haploid chromosome. Clinically, β-thalassemia may occur in major (homozygous), intermediate (usually compound heterozygous), and minor (heterozygous) genetic forms. In β-thalassemia major (TM), the severe form of the disease, both alleles are affected and life-threatening anemia develops. If untreated, anemia leads to tissue hypoxia and resultant redox imbalance.5,6 TM patients require lifetime blood transfusions, which lead to iron overload. This pathological condition plays a crucial role in the generation of reactive oxygen species (ROS) that are capable of mediating the cellular macromolecules including deoxyribonucleic acid (DNA). Patients suffer from cumulative oxidative damage produced by iron and hypoxia.5,6 Research on sickle cell disease (SCD) and iron deficiency anemia (IDA) indicates elevated oxidative stress and DNA damage. SCD, which is another hemoglobinopathy, similar to thalassemia, is linked to persistent inflammation and oxidative stress, resulting in increased DNA damage.7,8 Deficiency of iron, like its overload, although exhibiting less oxidative stress compared to thalassemia or SCD, may still induce genomic instability.9,10 Similar complications, to a lesser degree, develop in β-thalassemia intermedia. Thalassemia minor (Tm) is the less-severe form of the disorder and is usually expressed as only mild anemia with no clinical disability.11The main goal in the detection of Tm in individuals is the opportunity for genetic counseling. However, the potential clinical significance of Tm in some health fields, including DNA damage, has been described.12-18 There is extensive knowledge about DNA damage in patients with β-thalassemia; however, DNA damage in heterozygotes remains unclear. β-thalassemia is common and ascertaining DNA damage status in β-Tm individuals, the most frequent form, is important worldwide. Determination of potential DNA damage during childhood, known as a critical step in the initiation of cancer, is necessary to enable the regulation of living conditions for those individuals.
DNA damage refers to alterations in DNA structure that may lead to loss of genome integrity.19,20 Genotoxicity has been shown to be associated with radiation, cigarette smoking and environmental tobacco smoke (ETS) exposure, viral infections, vitamin deficiencies, and some genetic disorders (e.g., ataxia telangiectasia, Bloom’s syndrome).21-23 Oxidation of bases is one of the mechanisms of DNA damage.19,20 ROS generated by external agents or normal metabolic/cellular processes, as observed in thalassemia, have a genotoxic effect via oxidation reaction leading to DNA strand breaks.24-27 Among genotoxicity assays, the comet assay is widely used for the detection of DNA damage in several clinical conditions, including childhood diseases.28-31 Comet assay can directly assess the damage at a single cell level.
The aim of this study was to assess, for the first time, DNA damage by comet assay in a pediatric population with β-Tm using several biochemical parameters including oxidative status tests.
Materials and Methods
Subjects
The present study, assessed DNA damage in β-Tm. 142 children (73 males, 69 females; age: 6.0±3.6 years (mean±SD), range: 1-17 years) with β-Tm from the Pediatric Hematology Department of Gazi University Medical Faculty were recruited between July 1, 2014 and December 31, 2015. β-Tm was diagnosed based on red cell indices on complete blood count (CBC) and hemoglobin (Hb)A2 level in hemoglobin electrophoresis; microcytosis (mean corpuscular volume [MCV]<80 fL), hypochromia (mean corpuscular hemoglobin [MCH]<25 pg) and erythrocytosis (red blood cells [RBC]>5x106/L) on CBC, with normal body iron status, and simultaneously increased HbA2 level (≥3.5%) were consistent with the diagnosis. In some individuals with similar results but normal HbA2 levels, the diagnosis was achieved by parental study and molecular analysis of the β-globin gene. As controls, 113 children (50 males, 63 females; age: 8.2±3.4 years, range: 1-17 years) were enrolled. A segment of the control group was formed from healthy siblings of the β-Tm subjects (n=34, 30% of the controls). For those subjects with no healthy sibling, age- and gender-matched healthy children (n=79, 70% of the controls) were enrolled as their controls. Children with infections, a chronic or genetic disorder, taking any medications, or with a history of blood transfusion for any reason were not included. This study was approved by the ethics committee of Gazi University Faculty of Medicine (No: 162, Date: 24.03.2014) and supported by Gazi University Faculty of Medicine Research Fund (DA, 01/2014-19). The parents of the children were informed about the study and all gave informed consent prior to their children’s involvement. Detailed questionnaire forms were completed by the parents, and included age, height, weight, education, cigarette smoking and ETS exposure, recent diagnostic X-ray examination (yes/no; within 3 months prior to the sampling), use of vitamins (e.g., multivitamin, folate, vitamins B and C), viral infections (e.g., varicella, mumps) over the previous 2 years, recent vaccination in the previous year, and physical activity of the children.
Biological sampling
Biological sampling for both the patient and control groups was completed simultaneously between July 2014 and December 2015. Blood samples were collected by two certified phlebotomists by venipuncture to use in the analysis of hematological parameters, biochemical parameters, oxidative status tests, and DNA damage endpoint. In all subjects, hematological parameters of CBC, body iron status (iron, iron binding capacity, and ferritin levels), and hemoglobin electrophoresis as routine thalassemia diagnostic tests and biochemical parameters of serum vitamin B12, folate and C-reactive protein (CRP) were studied. Automated blood counts were performed using a Coulter (LH 780 Hematology Analyzer by Beckman Coulter), and all samples were analyzed on the Bio-Rad Variant TM II high performance liquid chromatography (HPLC) system. Remaining biochemical parameters were analyzed using a hormone analyzer and chemiluminescent immunoassay (Immulite 1000 apparatus [Siemens Health Diagnostics, U.S.]). For analysis of total oxidative status (TOS) and total antioxidant capacity (TAC), as oxidative status parameters, serum was separated by centrifugation for 10 min at 4500 rpm and stored at -80°C until analysis. The levels of TOS and TAC were determined using a commercially available kit (Rel Assay Diagnostics, Gaziantep, Turkey). The results were expressed as mmol Trolox Eqv./L and H2O2 Eqv./L, respectively. The levels of vitamins C and E, as antioxidative capacity tests, were measured using commercially available kits (Human Vitamin C, VC ELISA Kit [Cat no: CSB-E08090h] and Human Vitamin E,VE ELISA Kit [Cat no: CSB-E07893h], respectively). The results were expressed as ng/mL and mmol/mL, respectively. Blood samples in heparin-containing tubes were delivered to the laboratory within the same day for genotoxicity testing by comet assay.
Comet assay
The alkaline comet assay was performed on the day of sampling according to Singh et al. with slight modifications.32 Lymphocytes from 2 mL heparinized whole blood were isolated by density gradient centrifugation using Biocoll (AG Biochrom, Berlin, Germany). Lymphocytes were suspended in 100 μL of 0.65% low melting-point agarose (LMA) at 37°C, and 200 μL of mixture was layered onto a microscope slide precoated with 0.65% high melting–point agarose (HMA). The slide was immediately covered with a coverslip, and the agarose was allowed to solidify for 5 min on ice. Two gels were prepared for each sample. After removal of coverslips, the slides were immersed in light-protected cold lysing solution (89% lysing buffer [2.5 M NaCl, 0.1 M Na2EDTA, 10 mM Tris-HCl; pH 10], 1% Triton X-100, 10% dimethylsulfoxide) overnight at 4°C for lysis. Slides were pretreated for 20 min in freshly prepared electrophoresis buffer (0.3 M NaOH, 1 mM Na2EDTA: pH 13) for unwinding, then exposed to 25 V and 300 mA for 20 min at 4°C (Thermo EC250-90). The slides were neutralized three times for 5 min in the neutralizing buffer (0.4 M Tris-HCl: pH 7.5). The gels were then stained with 50 μL ethidium bromide (20 μg/mL), and 50 cells per slide were scored by use of the Comet Assay III image-analysis system (Perceptive Instruments, UK) with a fluorescence microscope (Zeiss Axioscope, Germany). Tail intensity (TI) (percent DNA in the tail) was chosen as the measure of DNA damage.
All parameters were compared between the patient and control groups. The patient group was further divided into age subgroups (<5 years [n=68, 48%], 5-10 years [n=60, 42%], >10 years [n=14, 10%]), and TI values were compared within these subgroups. A similar analysis was conducted for the control group.
Statistical analysis
The Statistical Package for the Social Sciences version 22.0 for Windows (SPSS, Inc., Chicago, IL, USA) was used in the analysis of the data. Receiver operating characteristic (ROC) curve analysis was used for determining the optimal cut-off value of age for discrimination between the β-Tm and control groups. Independent samples t-test was used to compare two independent groups (β-Tm and controls) for normally distributed variables, and Mann–Whitney U test was used for comparison of non-normally distributed variables. Pearson chi-square test was used to compare the two groups (β-Tm and controls) for differences in categorical variables. Arithmetic mean, standard deviation, median, and minimum-maximum values were given as descriptive statistics for quantitative data. Qualitative data were summarized using frequency and percentages. A p value of less than 0.05 was considered to indicate a statistically significant difference.
Results
Some general characteristics of the study population are shown in Table I. The groups were similar according to gender, body mass index (BMI), ETS exposure, viral infections over the previous 2 years, the frequency of use of vitamins, vaccination in the year prior, and X-ray exposure in 3 months prior (p>0.05 for all, Table I). Although the age range was similar, the mean age in the control group was found to be higher than that in the β-Tm group (8.2±3.4 versus 6.0±3.6 years, respectively) (p<0.05, Table I). The β-Tm group was further divided into 2 age groups according to ROC curve (≤7 years and >7 years) to determine any potential confounding effect of age on the baseline parameters. However, comparison of the age groups showed similar results, excluding an effect of age on these parameters.
| BMI: body mass index, ETS: enviromental tobacco smoke, SD: standard deviation. For comparisons, Mann Whitney U test, Pearson chi square and Fisher’s excact tests were used. Differences were accepted as statistically significant at a p value of <0.05. | |||
| Table I. General characteristics of the patient and control groups. | |||
| β-thalassemia minor | Control | P value | |
| Age (mean±SD) | 6.0±3.6 | 8.2±3.4 | <0.05 |
| Gender | |||
| Male, n (%) | 73.0 (51.4) | 50.0 (44.2) | >0.05 |
| Female, n (%) | 69.0 (48.6) | 63.0 (55.8) | >0.05 |
| BMI (mean±SD) | 17.1±3.3 | 17.2±3.2 | >0.05 |
| ETS exposure, n (%) | 25.0 (17.6) | 24.0 (21.2) | >0.05 |
| Viral infection, n (%) | 7.0 (5.0) | 6.0 (5.3) | >0.05 |
| Use of vitamin, n (%) | 8.0 (5.7) | 3.0 (2.7) | >0.05 |
| Vaccination, n (%) | 55.0 (39.0) | 26.0 (23.0) | >0.05 |
| X-ray exposure, n (%) | 29.0 (20.4) | 24.0 (21.2) | >0.05 |
Hematological and biochemical parameters of the groups are shown in Table II. Red cell indices of Hb, MCV, MCH, red cell distribution width (RDW), RBC and HbA2 level were significantly different between healthy controls and β-Tm children (p<0.05 for all, Table II). With regard to biochemical parameters including serum iron, transferrin saturation, serum ferritin, vitamin B12, folate, and CRP, β-Tm children and controls were similar (p>0.05 for all, Table II). According to oxidative status tests of TOS, TAC, and serum levels of vitamins C and E, the β-Tm group and control group were similar (p>0.05 for all, Table II).
| Hb: hemoglobin, MCV: mean corpuscular volume, MCH: mean corpuscular hemoglobin, RBC: red blood cell, RDW: red celrl distribution width, TAC: total antioxidant capacity, TOS: total oxidative status. Independent samples t-test was used to compare two independent groups that are β-thalassemia minor and control for serum iron, and Mann–Whitney U test was used for comparison for other variables. | |||||
| Table II. Descriptive statistics of hematological, biochemical, and oxidative status parameters in the patient and control groups. | |||||
| β-thalassemia minor | Control | P value | |||
| Median | (min-max) | Median | (min-max) | ||
| Hb (g/dL) | 11.6 | (8.1-14.7) | 13.0 | (10.2-17.0) | <0.001 |
| MCV (fL) | 65.0 | (53.4-79.8) | 83.0 | (57.9-89.7) | <0.001 |
| RBC (x106/L) | 5.5 | (4.4-7.1) | 4.7 | (4.0-6.2) | <0.001 |
| RDW (%) | 15.8 | (12.2-30.9) | 13.2 | (11.8-16.4) | <0.001 |
| MCH (pg) | 20.8 | (16.2-27.2) | 28.0 | (18.4-30.1) | <0.001 |
| Hb A2 (%) | 3.5 | (2.1-8.7) | 3.0 | (2.0-6.0) | <0.001 |
| Ferritin (ng/mL) | 33.4 | (7.8-223.7) | 34.5 | (10.4-149.3) | >0.05 |
| Transferrin saturation (%) | 26.3 | (3.4-104.2) | 26.8 | (6.8-106.1) | >0.05 |
| C-reactive protein (mg/L) | 1.9 | (0.3-12.7) | 2.0 | (1.0-16.6) | >0.05 |
| Vitamin B12 (pg/mL) | 396.0 | (117.3-1208.0) | 364.5 | (166.2-1240.0) | >0.05 |
| Folate (ng/mL) | 11.2 | (4.9-80.0) | 11.2 | (4.9-48.7) | >0.05 |
| TAC (mmol/L) | 2.2 | (0.2-4.0) | 2.2 | (0.2-4.2) | >0.05 |
| TOS (µmol/L) | 6.4 | (1.3-51.1) | 6.6 | (0.3-67.0) | >0.05 |
| Vitamin C (ng/mL) | 63.2 | (6.4-313.9) | 69.4 | (9.7-312.1) | >0.05 |
| Vitamin E (nmol/ml) | 30.9 | (10.3-130.3) | 37.7 | (10.4-128.9) | >0.05 |
| Aritmetic Mean | Std. deviation | Aritmetic Mean | Std. deviation | ||
| Iron (µg/dL) | 77.4 | 30.6 | 85.2 | 32.5 | >0.05 |
In the comet assay, TI showed no statistically significant difference between the β-Tm (n=142) and control groups (n=113) (median [min-max], 6.5 [2.6-31.8] versus 6.7 [1.7-40.9], respectively) (p=0.551, Fig. 1A). Similarly, TI was not significantly different between the β-Tm children (n=34) and their healthy siblings (n=34) (median [min-max]; 5.8 [2.6-11.3] versus 6.2 [2.9-12.4], respectively) (p=0.655, Fig. 1B). After further dividing the β-Tm group into three subgroups (<5 years [n=68, 48%], 5-10 years [n=60, 42%], >10 years [n=14, 10%]), TI was not significantly different among age subgroups, but a gradual increase by age was observed (median [min-max]; 6.1 [2.6-12.0], 7.1 [2.8-31.8], and 7.3 [4.0-11.9], respectively) (p=0.064 for all) (Fig. 1C). A similar increase was observed when the control group was divided into age subgroups, and this increase was also not statistically significant (median [min-max]; 6.2 [1.7-9.0], 6.3 [2.8-40.9], and 6.9 [3.1-12.3], respectively) (p >0.05 for all). The increased rate of TI in β-Tm group was higher than that in the control group.
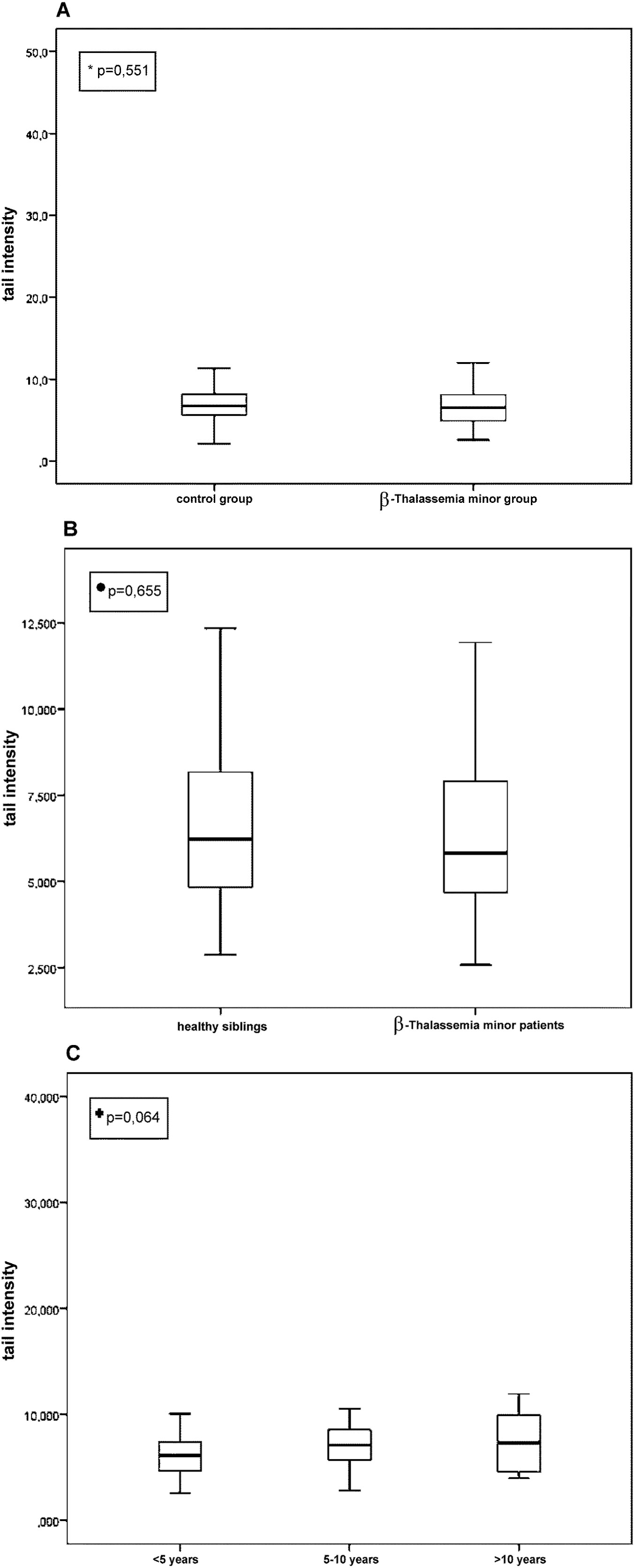
• β-Thalassemia minor patients (n=34) vs their healthy siblings (n=34).
+Among age groups of β-thalassemia minor children (<5 years [n=68, 48%), 5-10 years [n=60, 42%], >10 years [n=14, 10%]).
Hb levels did not exhibit a significant correlation with TI or oxidative status parameters (TOS, TAC, vitamins C and E) (p>0.05 for all).
Discussion
Hemoglobinopathies, including thalassemia and SCD, are genetic disorders affecting hemoglobin structure or synthesis. In severe forms, such as transfusion-dependent thalassemia, chronic hypoxia due to anemia and iron overload from transfusions contribute to oxidative stress and subsequent DNA damage. This cumulative oxidative stress is a key mechanism underlying genotoxicity in these conditions.5-7,10,33-39 Although β-Tm is the silent form of the disorder, β-Tm individuals have also been shown to be at a risk of oxidative stress generated from unpaired globin chains.40-43 In addition, lower antioxidative capacity has been shown in heterozygotes.44,45 Though the oxidative stress-related DNA status in heterozygotes is as yet unknown. By investigating DNA damage in children with β-Tm, the present study might help enlighten this. To our knowldge, to date only two genotoxicity studies have examined DNA damage status in β-Tm. In both studies, adult subjects were assessed, the sample size was small, and none or only a few biochemical parameters were studied. In the study by Al-Sweedan et al., for example, only 18 individuals were assessed. No parameter in the study by Krishnaja et al. and only a single parameter in the study by Al-Sweedan et al. (8-hydroxy-2’-deoxyguanosine [8-OHdG] level in urine, as oxidative stress parameter) was studied.12,17 Our study was carried out in a large population of children enabling appropriate statistical analysis using comet assay, and was strengthened by the use of several biochemical parameters. In addition, unlike in the previous two studies, our control group included healthy siblings of the subjects. Comet assay is widely used for genotoxicity assessment in various disorders and particularly in transfusion-dependent thalassemia.5,6,28,32-37 However, to date, it has not been used for genotoxicity assessment in Tm individuals. Considering these factors together, our study design is unique, with no comparison in the literature.
In this study, children with similar demographic features, socioeconomic conditions and habits were enrolled in the β-Tm and control groups. Although the mean age was found to be higher in the control group, further evaluation after controlling for the effect of age on other variables revealed no additional influennce on the results. It can, therefore, be said that the only distinctive feature between the patient and control groups was the presence of β-Tm. As a natural consequence, erythrocyte indices and HbA2 levels were significantly different between the groups, which also served the purpose of distinguishing the heterozygotes and healthy children. In children with β-Tm, a disorder characterized with hypochromic microcytic anemia, red cell indices of Hb, MCV and MCH were lower, and RBC and HbA2 levels, as expected, were higher.
Iron, folate and vitamin B12 have a povital role in maintaining healthy cell division and DNA synthesis. Deficiencies of these micronutrients inhibit purine and thymidylate synthesis.46,47 Impaired DNA synthesis can cause genomic instability, some of which may give rise to cancer. Results of studies investigating the levels of serum iron, folate, and vitamin B12 in heterozygotes have been inconsistent.44,48-51 Despite their certain effect on DNA synthesis and uncertain levels in heterozygotes, these micronutrients were not measured in the two previous studies investigating DNA damage in β-Tm. In our study, the levels of folate and vitamin B12 were assessed and found to be within normal limits. In all studies investigating DNA damage in patients with transfusion-dependent thalassemia, iron was measured because of its genotoxic effect when accumulated.5,6 Our study population, however, consisted of subjects with transfusion-free Tm. We measured serum iron as part of the diagnostic tests and because its deficiency might affect DNA synthesis. Our results with regard to all parameters of body iron status were similar with those of the controls and were within normal limits. The results for CRP, a marker for inflammation and infection commonly used in genotoxicity studies, were within normal limits and similar to those of the controls.52,53
Oxidative stress plays a pathological role in the development of various diseases, including cancer. Systemic oxidative stress results from an imbalance between oxidant derivatives production and antioxidants defenses. In β-Tm, there is an unbalanced globin chain synthesis, which results in excess α-globin chains in erythrocytes. This has been shown to generate ROS, which put the patients at a high risk of oxidative stress.40-42,44,45 In addition, it has been shown that heterozygotes have increased non-heme iron content of erythrocytes.54 Non-heme iron can inhibit the action of various cytoplasmic enzymes, thereby altering cellular homeostasis. Thus, the cell experiences oxidative stress and possibly significant protein degradation and lipid peroxidation in the cell membrane.55 Increased oxidative stress in heterozygotes may hypothetically be exacerbated by a decreased level of natural antioxidants, which was previously documented.37 Antioxidant defense can be evaluated by measurement of either individual antioxidant levels in cell and serum or TAC. In our study, oxidative stress was double-checked by testing both oxidants (measured as TOS) and antioxidants defenses (by measurement of the individual antioxidant levels of vitamins C and E in serum and also the TAC), and the results were found to be similar in heterozygotes and healthy controls. Those measurements were not taken in the two previous studies. In the study by Al-Sweedan et al., the observation of increased DNA damage was speculated to be related with the lower serum levels of these vitamins in heterozygotes; however, vitamin levels in their study were estimated based on literature knowledge and not measured directly.17 In our study, serum levels of vitamins C and E were measured and were not found to be significantly different from those in controls, probably due to our selection of the subjects and controls from the same environment and/or family (Table II).
Genotoxicity refers to all types of alterations in DNA sequence, and genotoxic events have been known to be a critical step in the initiation of cancer.21,25,27 The assessment of cancer risk using genotoxicity assays is of utmost importance. Patients with TM and SCD have an increased risk of cancer compared to the general population. While the underlying mechanisms of cancer development in these patients are not fully understood and are likely multifactorial, disease-related DNA damage is a highly probable contributing factor.56-60 Among the various methods employed in the estimation of DNA damage, the comet assay used in our study is proven to be a relatively simple, effective and versatile tool, and is a validated biomarker assay.28,31,32 With regard to DNA damage, we found no significant difference between the β-Tm and control groups (Fig. 1A). Further, there was no significant difference in DNA damage between the β-Tm children and their healthy siblings (Fig. 1B). However, after further dividing the β-Tm group into three age subgroups, TI was found to gradually increase with age. The most striking finding was that the control group had a higher mean age compared to the β-Tm group, and healthy siblings had a higher mean age than β-Tm children. Moreover, the TI value increased with advancing age in all age subgroups of the control group. These findings suggest that age has an impact on the TI value. Interestingly, the age-related increase in TI value was more rapid in β-Tm children compared to the control group. Although carrier children initially had numerically lower TI values than controls, they increased more rapidly within the same age range, even surpassing control TI values. These results indicate that while age has an increasing effect on the TI value, environmental factors may exert a more pronounced influence in the context of β-Tm. This supports the notion that β-Tm children should be monitored at appropriate intervals throughout adulthood and protected against certain risks.
In the two previous studies, micronuclei frequency in lymphocytes and chromosomal aberrations, sister chromatid exchanges and 8-OHdG assays were used to investigate DNA damage in adult heterozygous subjects.12,17 Thus, we cannot compare our study with these except for the fact that subjects were β-Tm individuals. In both studies, increased DNA damage was found in heterozygotes compared to healthy controls. The most likely explanation is age. Although there were several disadvantages regarding their study design, particularly their small population size, it may be speculated that the age factor and its related conditions (e.g., cigarette smoking, alcohol consumption, and environmental and occupational factors) might have contributed to the potential genotoxicity in adult subjects with Tm and augmented it to a detectable level. In accordance, the division of our β-Tm group into three groups according to age revealed numerical differences in DNA damage among the subgroups, suggesting that reassessment beyond the pediatric age range is necessary. A more severe effect of some mutations might also be responsible for the different results.61
The micronucleus (MN) assay in a thesis study investigating DNA damage in children with β-Tm found similar levels of genotoxicity, cytotoxicity, and oxidative stress parameters compared to controls. However, β-Tm children had a higher frequency of MN values above the cut-off level (39.2% vs. 21.6% in controls, p<0.05).62 This result obtained with a method that shows DNA damage at the cytome level, as in the comet assay, suggests a potential effect of β-Tm on DNA damage and emphasizes the need for monitoring carrier children.
Our study benefits from a substantial sample size and the evaluation of multiple factors linked to DNA damage. Additionally, we included a significant number of unaffected siblings in the control group. However, we were unable to measure intracellular vitamin levels and did not follow up on TI parameters in adulthood, which are our limitations.
In conclusion, our study, the first to our knowledge to investigate DNA damage in a large pediatric population with β-Tm using the comet assay, found no significant differences in DNA damage levels between β-Tm patients and healthy controls, nor between siblings with and without β-Tm. However, TI, a marker of DNA damage, increased more rapidly with age in β-Tm patients compared to healthy controls. These findings suggest that regular monitoring of DNA damage in β-Tm patients throughout childhood and adulthood, along with examining the relationship between TI and age-related risk factors, could provide valuable insights. Future studies employing diverse genotoxicity testing methods may help to further clarify the status of DNA damage in β-thalassemia.
Ethical approval
This study was approved by the Ethics Board of Gazi University Faculty of Medicine (No: 162, Date: 24.03.2014).
Source of funding
The authors declare that the study is supported/funded by Gazi University Research Fund, grant number: DA, 01/2014-19.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Cao A, Galanello R. Beta-thalassemia. Genet Med 2010; 12: 61-76. https://doi.org/10.1097/GIM.0b013e3181cd68ed
- Higgs DR. Gene regulation in hematopoiesis: new lessons from thalassemia. Hematology Am Soc Hematol Educ Program 2004; 2004: 1-13. https://doi.org/10.1182/asheducation-2004.1.1
- Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. Lancet 2012; 379: 373-383. https://doi.org/10.1016/S0140-6736(11)60283-3
- Marengo-Rowe AJ. The thalassemias and related disorders. Proc (Bayl Univ Med Cent) 2007; 20: 27-31. https://doi.org/10.1080/08998280.2007.11928230
- Ferro E, Visalli G, Civa R, et al. Oxidative damage and genotoxicity biomarkers in transfused and untransfused thalassemic subjects. Free Radic Biol Med 2012; 53: 1829-1837. https://doi.org/10.1016/j.freeradbiomed.2012.08.592
- Sagar CS, Kumar R, Sharma DC, Kishor P. DNA damage: beta zero versus beta plus thalassemia. Ann Hum Biol 2015; 42: 585-588. https://doi.org/10.3109/03014460.2014.990921
- Biswal S, Rizwan H, Pal S, Sabnam S, Parida P, Pal A. Oxidative stress, antioxidant capacity, biomolecule damage, and inflammation symptoms of sickle cell disease in children. Hematology 2019; 24: 1-9. https://doi.org/10.1080/10245332.2018.1498441
- Aslan M, Horoz M, Kocyigit A, et al. Lymphocyte DNA damage and oxidative stress in patients with iron deficiency anemia. Mutat Res 2006; 601: 144-149. https://doi.org/10.1016/j.mrfmmm.2006.06.013
- Esen Ağar B, Akarsu S, Aydin S. The effect of iron deficiency anemia and different treatment methods on DNA damage: 8-hydroxy-2-deoxyguanosine level. Glob Pediatr Health 2021; 8: 2333794X211041337. https://doi.org/10.1177/2333794X211041337
- Queiroz RF, Lima ES. Oxidative stress in sickle cell disease. Rev Bras Hematol Hemoter 2013; 35: 16-17. https://doi.org/10.5581/1516-8484.20130008
- Clarke GM, Higgins TN. Laboratory investigation of hemoglobinopathies and thalassemias: review and update. Clin Chem 2000; 46: 1284-1290.
- Krishnaja AP, Sharma NK. Heterogeneity of chromosome damage in beta-thalassaemia traits. An evaluation of spontaneous and gamma-ray-induced micronuclei and chromosome aberrations in lymphocytes in vitro after G0 and G2 phase irradiation. Int J Radiat Biol 1994; 66: 29-39. https://doi.org/10.1080/09553009414550921
- Tong PC, Ng MC, Ho CS, et al. C-reactive protein and insulin resistance in subjects with thalassemia minor and a family history of diabetes. Diabetes Care 2002; 25: 1480-1481. https://doi.org/10.2337/diacare.25.8.1480
- Karimi M, Karamifar HA. Short stature in beta-thalassemia minor subjects. Med Sci Monit 2004; 10: CR603-CR605.
- Namazi MR. Minor thalassemia may be a risk factor for impulsiveness. Med Hypotheses 2003; 60: 335-336. https://doi.org/10.1016/s0306-9877(02)00398-5
- Palma-Carlos AG, Palma-Carlos ML, Costa AC. "Minor" hemoglobinopathies: a risk factor for asthma. Eur Ann Allergy Clin Immunol 2005; 37: 177-182.
- Al-Sweedan SA, Khabour O, Isam R. Genotoxicity assessment in patients with thalassemia minor. Mutat Res 2012; 744: 167-171. https://doi.org/10.1016/j.mrgentox.2012.02.010
- Graffeo L, Vitrano A, Scondotto S, et al. β-Thalassemia heterozygote state detrimentally affects health expectation. Eur J Intern Med 2018; 54: 76-80. https://doi.org/10.1016/j.ejim.2018.06.009
- Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet 2014; 15: 585-598. https://doi.org/10.1038/nrg3729
- Khalil HS, Tummala H, Hupp TR, Zhelev N. Pharmacological inhibition of ATM by KU55933 stimulates ATM transcription. Exp Biol Med (Maywood) 2012; 237: 622-634. https://doi.org/10.1258/ebm.2012.011378
- Bonassi S, Znaor A, Ceppi M, et al. An increased micronucleus frequency in peripheral blood lymphocytes predicts the risk of cancer in humans. Carcinogenesis 2007; 28: 625-631. https://doi.org/10.1093/carcin/bgl177
- Bonassi S, Znaor A, Norppa H, Hagmar L. Chromosomal aberrations and risk of cancer in humans: an epidemiologic perspective. Cytogenet Genome Res 2004; 104: 376-382. https://doi.org/10.1159/000077519
- Fenech M. Chromosomal biomarkers of genomic instability relevant to cancer. Drug Discov Today 2002; 7: 1128-1137. https://doi.org/10.1016/s1359-6446(02)02502-3
- Cadet J, Wagner JR. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb Perspect Biol 2013; 5: a012559. https://doi.org/10.1101/cshperspect.a012559
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 2003; 17: 1195-1214. https://doi.org/10.1096/fj.02-0752rev
- Jena NR. DNA damage by reactive species: mechanisms, mutation and repair. J Biosci 2012; 37: 503-517. https://doi.org/10.1007/s12038-012-9218-2
- Kryston TB, Georgiev AB, Pissis P, Georgakilas AG. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat Res 2011; 711: 193-201. https://doi.org/10.1016/j.mrfmmm.2010.12.016
- Aykanat B, Demircigil GC, Fidan K, et al. Basal damage and oxidative DNA damage in children with chronic kidney disease measured by use of the comet assay. Mutat Res 2011; 725: 22-28. https://doi.org/10.1016/j.mrgentox.2011.07.005
- Bajpayee M, Kumar A, Dhawan A. The comet assay: assessment of in vitro and in vivo DNA damage. Methods Mol Biol 2013; 1044: 325-345. https://doi.org/10.1007/978-1-62703-529-3_17
- Collins AR. The comet assay for DNA damage and repair: principles, applications, and limitations. Mol Biotechnol 2004; 26: 249-261. https://doi.org/10.1385/MB:26:3:249
- Gunasekarana V, Raj GV, Chand P. A comprehensive review on clinical applications of comet assay. J Clin Diagn Res 2015; 9: GE01-GE05. https://doi.org/10.7860/JCDR/2015/12062.5622
- Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res 1988; 175: 184-191. https://doi.org/10.1016/0014-4827(88)90265-0
- Anderson D, Dhawan A, Yardley-Jones A, Ioannides C, Webb J. Effect of antioxidant flavonoids and a food mutagen on lymphocytes of a thalassemia patient without chelation therapy in the comet assay. Teratog Carcinog Mutagen 2001; 21: 165-174.
- Ruf AA, Jerwood D, Webb J, Anderson D. Sensitivity of different thalassaemia genotypes to food mutagens in the comet assay. Teratog Carcinog Mutagen 2003;(Suppl 2): 83-91. https://doi.org/10.1002/tcm.10078
- Ruf AA, Webb J, Anderson D. Modulation by flavonoids of the effects of a food mutagen in different thalassaemia genotypes in the comet assay. Teratog Carcinog Mutagen 2003;(Suppl 2): 93-102. https://doi.org/10.1002/tcm.10083
- Anderson D, Yardley-Jones A, Hambly RJ, et al. Effects of iron salts and haemosiderin from a thalassaemia patient on oxygen radical damage as measured in the comet assay. Teratog Carcinog Mutagen 2000; 20: 11-26. https://doi.org/b8p565
- Anderson D, Yardley-Jones A, Vives-Bauza C, Chua-Anusorn W, Cole C, Webb J. Effect of iron salts, haemosiderins, and chelating agents on the lymphocytes of a thalassaemia patient without chelation therapy as measured in the comet assay. Teratog Carcinog Mutagen 2000; 20: 251-264. https://doi.org/ctpj9h
- Tarang A, Mozdarani H, Akbari MT. Frequency of background and radiation-induced apoptosis in leukocytes of individuals with alpha-thalassemia variants, assessed by the neutral comet assay. Hemoglobin 2009; 33: 247-257. https://doi.org/10.1080/03630260903039586
- Shaw J, Chakraborty A, Nag A, Chattopadyay A, Dasgupta AK, Bhattacharyya M. Intracellular iron overload leading to DNA damage of lymphocytes and immune dysfunction in thalassemia major patients. Eur J Haematol 2017; 99: 399-408. https://doi.org/10.1111/ejh.12936
- Adhiyanto C, Hattori Y, Yamashiro Y, et al. Oxidation status of β-thalassemia minor and Hb H disease, and its association with glycerol lysis time (GLT50). Hemoglobin 2014; 38: 169-172. https://doi.org/10.3109/03630269.2014.892884
- Gerli GC, Beretta L, Bianchi M, Pellegatta A, Agostoni A. Erythrocyte superoxide dismutase, catalase and glutathione peroxidase activities in beta-thalassaemia (major and minor). Scand J Haematol 1980; 25: 87-92. https://doi.org/10.1111/j.1600-0609.1981.tb01370.x
- Ondei Lde S, Estevao Ida F, Rocha MI, et al. Oxidative stress and antioxidant status in beta-thalassemia heterozygotes. Rev Bras Hematol Hemoter 2013; 35: 409-413. https://doi.org/10.5581/1516-8484.20130122
- Winterbourn CC, McGrath BM, Carrell RW. Reactions involving superoxide and normal and unstable haemoglobins. Biochem J 1976; 155: 493-502. https://doi.org/10.1042/bj1550493
- Castaldi G, Bagni B, Trotta F, Menegale G, Cavallini AR, Piffanelli A. Folic acid deficiency in beta-thalassaemia heterozygotes. Scand J Haematol 1983; 30: 125-129. https://doi.org/10.1111/j.1600-0609.1983.tb01456.x
- Livrea MA, Tesoriere L, Pintaudi AM, et al. Oxidative stress and antioxidant status in beta-thalassemia major: iron overload and depletion of lipid-soluble antioxidants. Blood 1996; 88: 3608-3614.
- Khabour OF, Soudah OA, Aaysh MH. Genotoxicity assessment in iron deficiency anemia patients using sister chromatid exchanges and chromosomal aberrations assays. Mutat Res 2013; 750: 72-76. https://doi.org/10.1016/j.mrgentox.2012.09.006
- Koury MJ, Ponka P. New insights into erythropoiesis: the roles of folate, vitamin B12, and iron. Annu Rev Nutr 2004; 24: 105-131. https://doi.org/10.1146/annurev.nutr.24.012003.132306
- Gallerani M, Cicognani I, Ballardini P, et al. Analysis of folate and vitamin B12 in beta thalassemia minor. Riv Eur Sci Med Farmacol 1990; 12: 247-250.
- Madan N, Sikka M, Sharma S, Rusia U. Haematological parameters and HbA2 levels in beta-thalassaemia trait with coincident iron deficiency. Indian J Pathol Microbiol 1998; 41: 309-313.
- Mehta BC, Pandya BG. Iron status of beta thalassemia carriers. Am J Hematol 1987; 24: 137-141. https://doi.org/10.1002/ajh.2830240204
- Silva AE, Varella-Garcia M. Plasma folate and vitamin B12 levels in beta-thalassemia heterozygotes. Braz J Med Biol Res 1989; 22: 1225-1228.
- Chumduri C, Gurumurthy RK, Zietlow R, Meyer TF. Subversion of host genome integrity by bacterial pathogens. Nat Rev Mol Cell Biol 2016; 17: 659-673. https://doi.org/10.1038/nrm.2016.100
- Pálmai-Pallag T, Bachrati CZ. Inflammation-induced DNA damage and damage-induced inflammation: a vicious cycle. Microbes Infect 2014; 16: 822-832. https://doi.org/10.1016/j.micinf.2014.10.001
- Musallam KM, Rivella S, Vichinsky E, Rachmilewitz EA. Non-transfusion-dependent thalassemias. Haematologica 2013; 98: 833-844. https://doi.org/10.3324/haematol.2012.066845
- Freikman I, Amer J, Cohen JS, Ringel I, Fibach E. Oxidative stress causes membrane phospholipid rearrangement and shedding from RBC membranes-an NMR study. Biochim Biophys Acta 2008; 1778: 2388-2394. https://doi.org/10.1016/j.bbamem.2008.06.008
- Alavi S, Safari A, Sadeghi E, Amiri S. Hematological malignancies complicating β-thalassemia syndromes: a single center experience. Blood Res 2013; 48: 149-151. https://doi.org/10.5045/br.2013.48.2.149
- Halawi R, Cappellini MD, Taher A. A higher prevalence of hematologic malignancies in patients with thalassemia: background and culprits. Am J Hematol 2017; 92: 414-416. https://doi.org/10.1002/ajh.24682
- Hodroj MH, Bou-Fakhredin R, Nour-Eldine W, Noureldine HA, Noureldine MHA, Taher AT. Thalassemia and malignancy: an emerging concern? Blood Rev 2019; 37: 100585. https://doi.org/10.1016/j.blre.2019.06.002
- Schultz WH, Ware RE. Malignancy in patients with sickle cell disease. Am J Hematol 2003; 74: 249-253. https://doi.org/10.1002/ajh.10427
- Seminog OO, Ogunlaja OI, Yeates D, Goldacre MJ. Risk of individual malignant neoplasms in patients with sickle cell disease: English national record linkage study. J R Soc Med 2016; 109: 303-309. https://doi.org/10.1177/0141076816651037
- Shekhar HU. Comment on: oxidative stress and antioxidant status in beta-thalassemia heterozygotes. Rev Bras Hematol Hemoter 2013; 35: 385-386. https://doi.org/10.5581/1516-8484.20130125
- Özel Babacanoğlu E, Emerce E, Kargın Menderes D, Arslan U, Aslan D, Çakmak G. Genotoxicity assessment of children with β-thalassemia minor by use of micronucleus assay in peripheral blood lymphocytes. 2nd International Gazi Pharma Symposium Series (GPSS-2017); Oct 11-13, 2017; Ankara, Türkiye.
Copyright and license
Copyright © 2025 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









