
Abstract
Background. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) variants are essential for determining eligibility for CFTR modulator drugs (CFTRms). In contrast to Europe and the USA, the treatment eligibility profile of cystic fibrosis (CF) patients in Türkiye is not known. In this study we aimed to determine the eligibility of CF patients in Türkiye for the CFTRms.
Methods. The Cystic Fibrosis Registry of Türkiye (CFrT) data was used to determine the age of patients in the year 2021 and the genetic variants they were carrying. Age- and CFTR-variant appropriate modulator therapies were determined using the Vertex® algorithm.
Results. Among a total of 1930 registered patients, CTFR gene analysis was performed on a total of 1841 (95.4%) patients. Mutations were detected in one allele in 10.7% (198 patients), and in both alleles in 79% (1455 patients) of patients. A total of 855 patients (51.7% for whom at least 1 mutation was detected) were eligible for the drugs. The most appropriate drug among genotyped patients was found to be elexacaftor/tezacaftor/ivacaftor for 486 patients (26.4%), followed by ivacaftor for 327 patients (17.7%) and lumacaftor/ivacaftor for 42 patients (2%).
Conclusions. Only half of patients registered in CFrT were eligible for CFTRms, which is a significant difference from the CFTR variant profile seen in USA and Europe. However, access to treatment is hampered for some patients whose genes are not analysed. Further studies in CF populations, where rare mutations are relatively more common, will contribute to the field of CFTR modulator treatments for such rare mutations.
Keywords: cystic fibrosis, cystic fibrosis transmembrane conductance regulator, CFrT registry, modulator, treatment
Introduction
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) modulator drugs (CFTRms) are genome-specific drugs that target the malfunctioning or defective protein produced by the CFTR gene.1 Although cystic fibrosis (CF) affects all races, the distribution of CFTR variants varies by race and geographic region.2,3 The F508del variant accounts for the majority genetic profile in the United States of America (USA) and European CF registries, and can on its own, sufficiently cover 90% and 85% of the CF population eligible for CFTRms, respectively.
Türkiye is a country located in southeastern Europe and western Asia. The peculiarity of this region is that it is home to different ethnic origins. Thus, its location causes a wide spectrum of CFTR variant diversity.4-7 Previous studies have shown that the most common CFTR variant, F508del, covers less than 30% of the Turkish CF population.4-7
Previous treatments for CF disease had only targeted symptoms, but CFTRms, which target the underlying problem, are now successfully used. With the introduction of these therapies, improvements in the health status of the CF cohort have been seen.8 However, CFTRm therapies are only effective in people with specific CFTR variants. There are four CFTRm drugs approved, which are ivacaftor (IVA), lumacaftor/ivacaftor (LUM/IVA), tezacaftor/ivacaftor (TEZ/IVA), and elexacaftor/tezacaftor/ivacaftor (ETI).9
Issues in accessing CFTRms in many countries including Türkiye, currently sets the main agenda of the CF population. In the USA and many countries in Europe, CFTRms are reimbursed, but Türkiye does not cover the costs of these treatments and their cost is far beyond what patients can individually afford. Some eligible patients have access to therapy through lawsuits, which involves a challenging process. The lack of knowledge of CFTR variants of the patients with CF is another important issue in the era of CFTRms, and limited access to genetic analysis hinders the knowledge of CFTRm eligibility. Profiling the CF population and their genetic characteristics can enable us to overcome the difficulties encountered in the era of CFTRms.
The purpose of this study was to identify the eligibility of CF patients for CFTRms registered in the Cystic Fibrosis Registry of Türkiye (CFrT). Also, suggestions on how to handle the problems are discussed.
Material and Methods
Study design and population
Cystic Fibrosis Registry of Turkiye (CFrT) is a web based patient registry which was established in 2007 by the Turkish Pediatric Respiratory Diseases and Cystic Fibrosis Society.10 Data of patients who fulfill the diagnostic criteria of CF are included by each center in a software program that is specifically developed for the CFrT. This registry consists of 25 demographic and 79 annually recorded data for each patient which includes genotyping, sweat test, nutrition, lung function, microbiology, treatments and complications.11
In this descriptive study, the data from patients with CF registered in the CFrT in the year 2021 were used. The ages and CFTR variants of the patients from 34 CF centers throughout the country were evaluated.
The decision of eligibility of CFTR variants for four modulator treatments (ETI, TEZ/IVA, LUM/IVA, IVA) was determined by using the ‘Vertextreatment finder’ (Finder) on the Vertex® website.12 The total number and percentages of ‘drugs offered by the Finder’ was determined. According to ‘Finder’ more than one drug might be offered to a patient. If more than one drug was recommended, the ‘most appropriate drug’ (MAD) was determined. This was defined as ‘the drug that could be primarily preferred’ depending on the literature and Institute for Clinical and Economy Review (ICER) evidence ratings on clinical effectiveness analysis, as follows:
A. For patients who were eligible for both TEZ/IVA and ETI, ETI was determined as MAD because of the clinical superiority of ETI treatment upon TEZ/IVA.13,14
B. For patients who were eligible for both LUM/IVA and ETI, MAD was determined according to each drug’s recommended starting age.
- For patients aged between 1-6 years, LUM/IVA was determined as MAD.
- For patients aged 6 years and above, ETI was determined as MAD, assuming the clinical effectiveness of ETI treatment for patients aged 6 years and above was superior to LUM/IVA.12
These assumptions regarding the clinical superiority and effectiveness of the drugs were made based on the literature and ICER evidence ratings.14 The use of ETI was approved for expanded use from the age of 12 years to age 6 years in 2021, and the use of LUM/IVA was approved for expanded use from the age of 2 years to age 1 year in 2022.9 We performed the evaluations according to the above regulations. The outcomes are presented as numbers and percentages for patients and eligible drugs.
The study was conducted in accordance with the ethical standards of the institutional research committee (Hacettepe University Ethics Board, date: 12 April 2007, reference number: HEK 07/16-2, Date: 5 June, 2018, reference number: GO 18/473-31) and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent was obtained from all patients or their parents/legal guardians.
Statistical analysis
Outcomes were the numbers and percentages of cases, the names of CFTR variants and the drugs for which the patients were eligible. Continous variables (age) were presented as mean ± standard deviation for normally distributed variables. Numbers and percentages (CFTR variants and CFTRms) were reported for categorical variables. Data were analysed using Statistical Package for the Social Sciences (SPSS) version 22.0 (SPSS Inc., Chicago, IL, USA).
Results
Demographic features
There were a total of 1948 patients registered in the CFrT in 2021, 1930 of whom were alive. Among the surviving patients, 1629 (84.4%) were children and adolescents, and 301 (15.6%) were adults. A total of 1841 (95.4%) surviving patients were found to have undergone CFTR genetic analysis. Of the surviving patients, 986 (53.5%) were male and 855 (46.4%) were female, with a mean age of 10.66±9.19 years. The number of patients who had not undergone genetic tests was 89 (4.6% of surviving patients).
Genetics
Among the 1841 patients who had been genotyped, two variants were identified in 1455 (79%) patients, only one variant was detected in 198 (10.7% ) patients, and no variants were detected in 188 (10.2%) patients (Fig. 1).
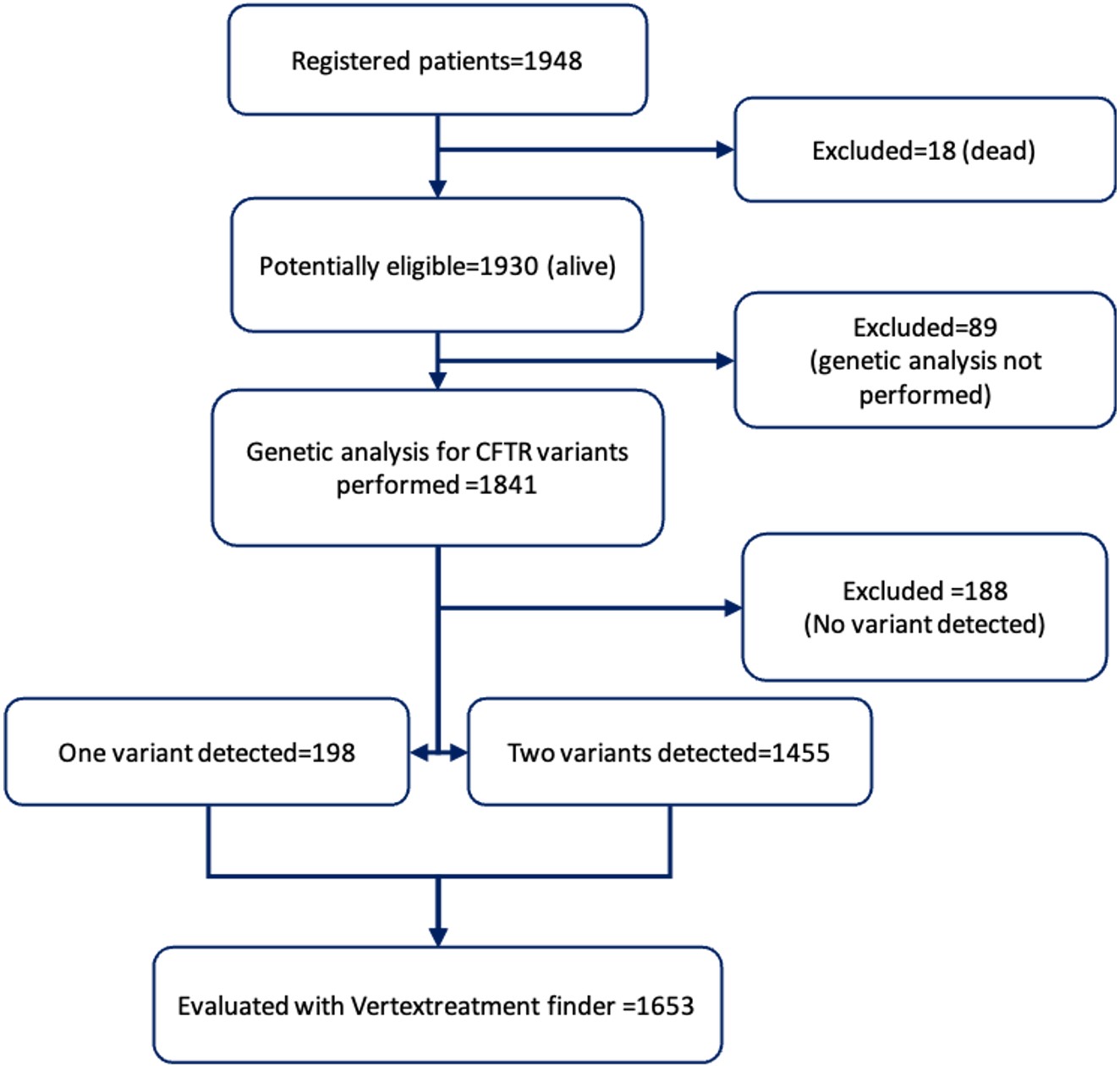
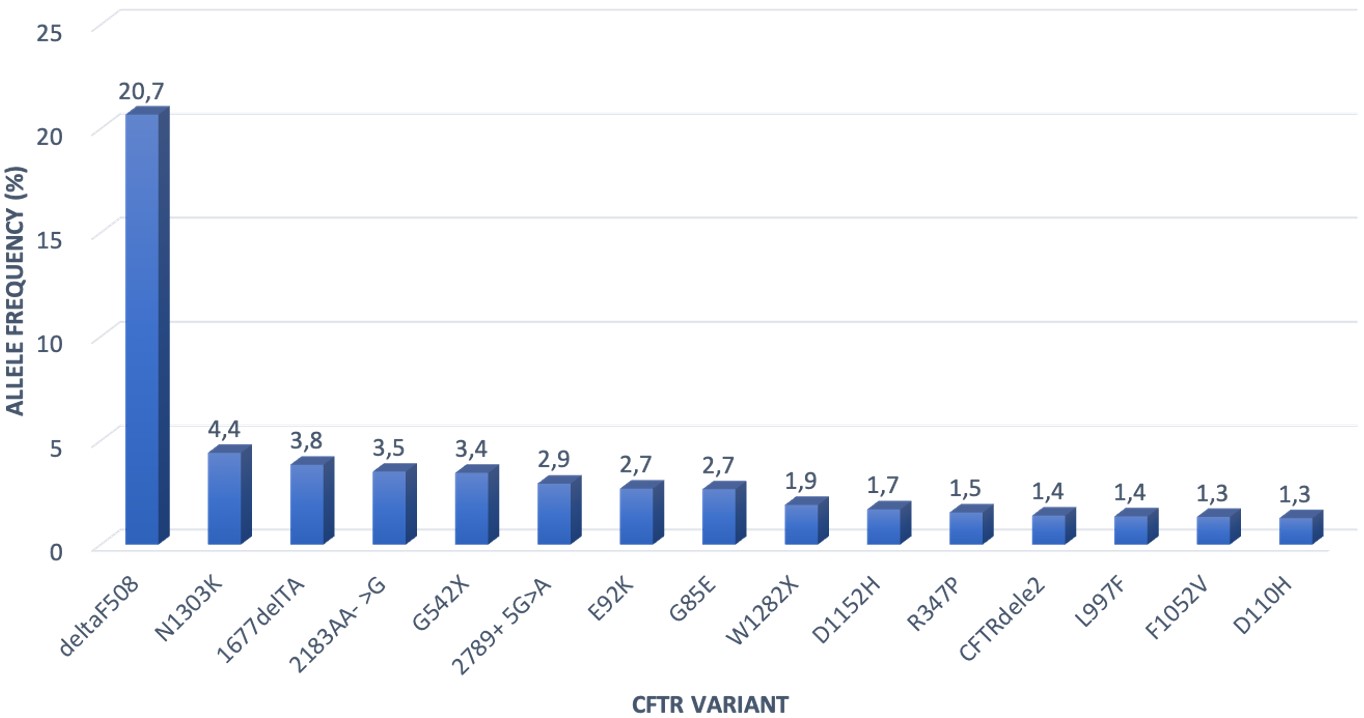
A total of 3,108 variants were observed in 1,653 surviving and genotyped patients, with 339 different variants. The most common variant was F508del in 692 alleles (22.2% of all alleles), followed by N1303K in 146 (4.7% of all) alleles and 1677delTA in 127 (4.1% of all) alleles.
Among the 1841 surviving patients who had been genotyped, 483 patients (25%) had the F508del variant on at least one allele. Two hundred patients were homozygous, and 283 patients were heterozygous for the F508del variant (comprising 10.8% and 15.3% of genotyped patients, respectively).
The most common 15 identified variants accounted for 58% of all alleles. Twenty different variants with a frequency of over 1% were identified. These 20 variants accounted for 63.7% of all alleles. The distribution of the most common 15 variants is given in Fig. 2.
Eligibility of the patients with CF for CFTRms
A total of 855 patients (51.7% of the patients for whom at least one allele was known) were eligible for any modulator drug, which constituted 44.3% of the 1930 total surviving patients, 46.4% of 1841 genotyped patients, and 51.7% of 1653 patients with at least one identified variant. The most common recommended drug was ETI (26.4% and 29.4%), followed by IVA (17.7% and 19.8%) and LUM/IVA (2.3% and 2.5%) among the genotyped patients and those with at least one identified variant, respectively. TEZ/IVA treatment was not the drug of choice for any patients (Table I).
| ETI: elexacaftor/tezacaftor/ivacaftor, IVA: ivacaftor, LUM/IVA: lumacaftor/ivacaftor, TEZ/IVA: tezacaftor/ivacaftor | ||||||
| Table I. The most appropriate drugs (MAD) for eligible patients by age | ||||||
| Modulator drugs | Age (years) | Number of eligible patients | Percentage among patients with at least one identified variant (N=1653 patients) (%) |
Percentage among genotyped patients (N=1841 patients) (%) |
Percentage among surviving patients (N=1930 patients) (%) |
|
| ETI | <18 | 369 | 29.4 | 26.4 | 25.1 | |
| ≥18 | 117 | |||||
| TEZ/IVA | All | 0 | 0 | 0 | 0 | |
| IVA | <18 | 267 | 19.8 | 17.7 | 16.9 | |
| ≥18 | 60 | |||||
| LUM/IVA | <18 | 42 | 2.5 | 2.3 | 2.17 | |
| ≥18 | 0 | |||||
| Total | <18 | 678 | 51.7 | 46.4 | 44.3 | |
| ≥18 | 177 | |||||
Among patients who had at least one identified allele, 43.7% of pediatric patients and 61.0% of adult patients were found to be eligible for a modulator drug. ETI was the most commonly recommended drug in both age groups.
‘Finder’ recommended a total of 1686 drugs to 855 patients. Among all offered drugs, 633 (37.5%) were ETI, 482 (28.6%) were TEZ/IVA, 375 (22.2%) were IVA, and 196 (11.6%) were LUM/IVA (Table II).
| ETI: elexacaftor/tezacaftor/ivacaftor, IVA: ivacaftor, LUM/IVA: lumacaftor/ivacaftor, TEZ/IVA: tezacaftor/ivacaftor | |
| Table II. The drugs offered by ‘Finder’ | |
| Modulator drugs | Number of patients offered for drug by 'Finder' (n) (percentage among all recommended drugs) (%) |
| ETI | 633 (37.5) |
| TEZ/IVA | 482 (28.6) |
| IVA | 375 (22.2) |
| LUM/IVA | 196 (11.6) |
| Total | 1686 |
It is noteworthy that TEZ/IVA treatment was offered for 482 patients by ‘Finder’ for eligible mutations, but was not identified as MAD for the patients (Table I and Table II).
Discussion
This study demonstrated that almost half of the registered patients were eligible for a CFTRms. The most common variant ‘F508del’ constituted 22.2% of all identified variants. ETI treatment was found to be the most appropriate drug common in 26.4% of genotyped patients. Genetic analysis of the CFTR gene was not performed in 4.6% of surviving patients.
There are some barriers regarding access to CFTRms in Türkiye, like in many other countries. The first step for revealing the obstacles that hinder knowledge of eligibility to CFTRms should be to determine the patient profile of the country. The data from the CFrT, which represents over 60% of the CF population in Türkiye, serves as valuable data for the purpose of drawing realistic conclusions regarding the eligibility status for the CFTRms in our country. Evaluation of the results showed many differences between European, American, and Turkish populations. Here, we would like to identify the genetic characteristics of our patients and highlight the issues that need to be solved.
The first difference between CF patients in Europe, the USA, and Türkiye that we established, was the prevalence of the variant F508del. The last annual reports of Cystic Fibrosis Foundation (CFF) and European Cystic Fibrosis Society (ECFS) patient registries showed that 85.5% and 80.3% of their genotyped patients had the F508del variant, respectively.8,15 In a 2018 study that evaluated the eligibility of patients in the CFrT from Türkiye, F508del was found in 34.6% of patients who were genotyped.16 In our study, the most common variant, F508del, accounted for 26.2% of surviving patients. Accordingly, it can be said that while a few CFTR variants are sufficient to provide eligibility for CFTRms in USA and Europe, the current qualifying CFTR variants for CFTRms only allow for less than 50% of our CF population to be eligible for such therapies.
Secondly, even though verification of CFTR variants is mandatory to identify a patient’s eligibility for CFTR modulators, CFTR variants of approximately 15% of our surviving patients were missing in our cohort. This group consisted of patients who did not undergo genetic testing (4.8%) and those whose mutation could not be detected (10.2%) even though genetic testing was performed. Both 2021 annual reports of the ECFS and CFF showed that 99.4% (50,849 and 48,814 patients) of the registered patients were genotyped.8,9 According to the ECFS 2021 report, Türkiye was one of the countries ranked at the top with unknown mutations.15 Nevertheless, the number of patients who were genotyped increased from 87.4% in 2017 to 95.4% in 2021 in patients in the CFrT.10,16 Despite the improvements in genotyping efforts over the years, we are still behind compared with Europe and USA. The success of genetic tests is important to find patients who qualify for CFTRms. No CFTR variant was identified in 25% and 19% of patients according to the 2017 and 2018 annual reports of the CFrT, respectively.10,16 In 2021, we still failed to detect mutations in 10% of cases.17 Since genetic analysis methods employed in the CF patients were not collected in the CFrT, it was not possible to draw any conclusions regarding analysis methods used. We speculate that the use of small CFTR panels may be the cause of these unknown mutations. Dayangaç-Erden et al.7 showed that the variant detection rate increased from 49.2% with DNA strip analysis to 76.7% with DNA sequence analysis, and CFTR panel use might not be sufficient to achieve the expected success in identifying the CFTR variants in our population. Expansion of DNA sequencing analysis, and multiplex ligation-dependent probe amplification (MLPA) throughout the country may help attain the goal of detection of CFTR variants. Also, efforts should be increased to ensure that genetic studies can be performed for all patients to identify eligibility for these treatments.
One of the reasons for the relatively low prevalence of drug eligibility in our population is the genetic diversity of the country. According to the worldwide analysis of CFTR variants, unlike the homogeneity of CFTR variants in central, northern, western, and northeastern European countries, it has been shown that Spain, Greece, Bulgaria, and Türkiye, which are defined as ‘gateway’ countries across Asia and Europe, show a widespread heterogeneous CFTR variant spectrum. Accordingly, whereas 10.2 variants per country constitute 78.9% of all variants in European countries, it has been reported that 25 variants constitute 84% of all variants in ‘gateway’ countries. A comparison of ‘gateway’ countries and others showed a statistically significant difference (p <0.001) in the extent of variants.18 Latter studies also support these results.4,6 Herein, we identified 339 different CFTR variants in surviving patients, and the most common 15 variants accounting for 63.7% of all the alleles. The conclusion here would be that high genetic diversity reduces the eligibility prevalence for CFTRms. Additionally, high genetic diversity may be indicative of a higher prevalence of rare CFTR variants that are not amenable to CFTRms. The effect of these drugs on many known mutations has not been studied yet. As highlighted by Fajac and Sermet, there may be CFTR variants that are not amenable to but may respond to CFTRms. Ways of assessing the effect of these drugs in people who carry these relatively few variants should be explored. In vitro studies showing its efficacy were sufficient for the United States Food and Drug Administration (FDA) to expand the extent of the mutations for IVA treatment in 2017. Similarly, it is important to find ways to evaluate drugs for this small population.19 When we looked at the number of eligible patients in the present study, 44.3% of surviving patients were eligible for any type of CFTRm treatment and ETI was the most commonly qualified drug (25% of genotyped patients). Recent data from CFF and ECFS registries showed that 90% and more than 80% of patients with CF were eligible for ETI treatment, respectively.8,9 Given the genetic profile of Türkiye, expanding the genetic coverage of drugs will enable more of our patients to benefit from these drugs.
Another issue that we would like to highlight is the challenges faced in accessing therapies in Türkiye. The main problems that impede access to the therapies are their cost and the reimbursement policies of the Social Security Institution (SSI) in Türkiye. The annual list prices of CFTRms are between $270,000 and $310,000.14 Considering that patients with CF must use these drug lifelong, it is not possible to afford them individually unless they are reimbursed by health insurance systems. Some of our patients can access treatment through lawsuits which is a very long, exhaustive and expensive process.
We agree with the idea put forward by Guo et al.20 who emphasized that efforts to access these treatments should be through global practices rather than individual efforts, and the prices should be reduced. Also, health systems should cover the costs, similarly to methods used in treatments such as HIV and tuberculosis.20
In addition, there are other mutations, which are not rare, in which the CFTR protein is not produced in more than ten percent. CFTRms are not effective for these mutations, which are nonsense mutations, frame-shift mutations, large deletions, insertions, and splice-site mutations. Preclinical studies are ongoing for these mutations. However, some issues need to be addressed immediately in the application to daily clinical practices.19
Our study has some limitations. First, this is a retrospective study, in which the data of the CFrT belonged only to the year 2021, thus the results do not show the current status of our population, but can only be a reflection of the present. The other limitation is that the type of the genetic testing methods of the CF centers was unknown. Because we did not know genetic analysis methods used, we could not evaluate the exact genetic status of all patients. Additionally, these results cannot be generalized to the whole country or worldwide. The power of our study is that CFrT data encompasses 60% of the CF population in our country, with data collected from a diverse range of geographical regions. This constitutes a significant proportion of the Turkish CF population. This study can lead each country to take action to determine the status of their patients.
Conclusions
The CFTR variant profile of Türkiye is very different from USA and most European countries. Approximately half of the patient population registered in CFrT was eligible for CFTRms. Nevertheless, the inability to perform CFTR gene analysis on some patients, even in small numbers, represents a barrier to their access to treatment. A determination of the prevalence of variants and genetic testing in the CF patient community may provide insight into barriers to drug access. Further studies on this subject in populations where rare mutations are relatively more common will contribute to the field of knowledge regarding CFTR modulator treatments for these rare mutations.
Ethical approval
This research was reviewed and approved by the institutional research committee (Hacettepe University Ethics Board, date: 12 April 2007, reference number: HEK 07/16-2, Date: June 5th, 2018, reference number: GO 18/473-31). Informed consent was obtained from all patients or their parents/legal guardians.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Pranke I, Golec A, Hinzpeter A, Edelman A, Sermet-Gaudelus I. Emerging therapeutic approaches for cystic fibrosis. From gene editing to personalized medicine. Front Pharmacol 2019; 10: 121. https://doi.org/10.3389/fphar.2019.00121
- McGarry ME, McColley SA. Cystic fibrosis patients of minority race and ethnicity less likely eligible for CFTR modulators based on CFTR genotype. Pediatr Pulmonol 2021; 56: 1496-1503. https://doi.org/10.1002/ppul.25285
- Grasemann H, Ratjen F. Cystic fibrosis. N Engl J Med 2023; 389: 1693-1707. https://doi.org/10.1056/NEJMra2216474
- Onay T, Topaloglu O, Zielenski J, et al. Analysis of the CFTR gene in Turkish cystic fibrosis patients: identification of three novel mutations (3172delAC, P1013L and M1028I). Hum Genet 1998; 102: 224-230. https://doi.org/10.1007/s004390050683
- Kılınç MO, Ninis VN, Dağlı E, et al. Highest heterogeneity for cystic fibrosis: 36 mutations account for 75% of all CF chromosomes in Turkish patients. Am J Med Genet 2002; 113: 250-257. https://doi.org/10.1002/ajmg.10721
- Atag E, Bas Ikizoglu N, Ergenekon AP, et al. Novel mutations and deletions in cystic fibrosis in a tertiary cystic fibrosis center in Istanbul. Pediatr Pulmonol 2019; 54: 743-750. https://doi.org/10.1002/ppul.24299
- Dayangaç-Erden D, Atalay M, Emiralioğlu N, et al. Mutations of the CFTR gene and novel variants in Turkish patients with cystic fibrosis: 24-years experience. Clin Chim Acta 2020; 510: 252-259. https://doi.org/10.1016/j.cca.2020.07.033
- Cystic Fibrosis Foundation. Patient Registry 2021 Annual Data Report. 2022. Available at: https://www.cff.org/sites/default/files/2021-11/Patient-Registry-Annual-Data-Report.pdf (Accessed on September 24, 2023).
- U.S. Food & Drug Administration. Available at: https://www.fda.gov (Accessed on September 24, 2023).
- Dogru D, Çakır E, Şişmanlar T, et al. Cystic fibrosis in Turkey: first data from the national registry. Pediatr Pulmonol 2020; 55: 541-548. https://doi.org/10.1002/ppul.24561
- Çocuk Solunum Yolu Hastalıkları ve Kistik Fibrozis Derneği. Ulusal Kistik Fibrozis Hasta Kayıt Sistemi. Available at: https://www.kistikfibrozisturkiye.org/hasta-kayit-sistemi/ (Accessed on September 24, 2023)
- Vertex. Find Treatment Options. Available at: https://www.vertextreatments.com (Accessed on September 24, 2023).
- Li Q, Liu S, Ma X, Yu J. Effectiveness and safety of cystic fibrosis transmembrane conductance regulator modulators in children with cystic fibrosis: a meta-analysis. Front Pediatr 2022; 10: 937250. https://doi.org/10.3389/fped.2022.937250
- Tice JA, Kuntz KM, Wherry K, et al. Modulator treatments for cystic fibrosis: effectiveness and value; final evidence report and meeting summary. Institute for Clinical and Economic Review; September 23, 2020. Available at: https://icer.org/wp-content/uploads/2020/08/ICER_CF_Final_Report_092320.pdf (Accessed on September 24, 2023).
- Zolin A, Orenti A, Jung A, et al. ECFSPR 2021 Annual Data Report. Available at: https://www.ecfs.eu/sites/default/files/Annual%20Report_2021_09Jun2023.pdf (Accessed on September 24, 2023).
- Çobanoğlu N, Özçelik U, Çakır E, et al. Patients eligible for modulator drugs: data from cystic fibrosis registry of Turkey. Pediatr Pulmonol 2020; 55: 2302-2306. https://doi.org/10.1002/ppul.24854
- Çocuk Solunum Yolu Hastalıkları ve Kistik Fibrozis Derneği. Ulusal Kistik Fibrozis Kayıt Sistemi 2021 Yılı Verileri. Available at: https://www.kistikfibrozisturkiye.org/wp-content/uploads/2022/11/UKKS-2021-raporu-2.pdf (Accessed on September 24, 2023).
- Bobadilla JL, Macek M, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations-correlation with incidence data and application to screening. Hum Mutat 2002; 19: 575-606. https://doi.org/10.1002/humu.10041
- Fajac I, Wainwright CE. New treatments targeting the basic defects in cystic fibrosis. Presse Med 2017; 46: e165-e175. https://doi.org/10.1016/j.lpm.2017.01.024
- Guo J, Garratt A, Hill A. Worldwide rates of diagnosis and effective treatment for cystic fibrosis. J Cyst Fibros 2022; 21: 456-462. https://doi.org/10.1016/j.jcf.2022.01.009
Copyright and license
Copyright © 2025 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









