
Graphical Abstract

Abstract
Objective. Phenylketonuria (PKU) is a prevalent inherited metabolic disorder, resulting from biallelic pathogenic variants in the PAH gene. This study aimed to assess the clinical characteristics of 1103 infants referred to a single center due to positive newborn screening (NBS) tests for PKU, providing insights into screening and diagnosis.
Methods. The health records of infants who were referred with suspicion of PKU through the Turkish national NBS program to a single referral center between January 2016 and January 2023 were retrospectively reviewed. The study analyzed demographic data, clinical findings, and diagnostic results from hospital records. Logistic regression analysis identified significant predictors of age at admission.
Results. This study highlights significant regional differences within Türkiye regarding DBS collection, result reporting, and age at admission. Significant delays in age at admission (expressed as median, [Q1-Q3]) were noted in the Eastern Anatolia (34 days [27-42]), Southeastern Anatolia [34 days (25-42)], and Black Sea regions [26 days (19-33)]. Out of the referred infants, 5.1% and 2.4% had transient tyrosinemia and transient hyperphenylalaninemia, respectively, and these transient conditions were more prevalent among neonates with a history of jaundice. Phenylalanine level was normal in 38.1% of the patients and was considered false positive. Among the 26 (2.36%) patients admitted after 90 days (late admissions), there were 2 PKU patients with untreated Phe levels >20 mg/dL (n=2). Among the 140 infants requiring treatment, 1.43% (n=2) were late admissions (>90 days). A history of PKU in the family and higher initial Phe levels were associated with earlier admissions.
Conclusion. This comprehensive analysis underscores the need to enhance NBS programs, particularly in regions with identified delays. Improving healthcare infrastructure, increasing awareness, and implementing targeted health policies are crucial for timely diagnosis and treatment. Future research should address regional disparities and optimize screening protocols to improve outcomes for affected infants.
Keywords: hyperphenylalaninemia, phenylketonuria, newborn screening, geographic disparities, delayed diagnosis
Introduction
Phenylketonuria (PKU) is one of the most common inherited metabolic disorders, caused by pathogenic variants in the PAH gene, which encodes the enzyme phenylalanine hydroxylase. This enzyme, which requires tetrahydrobiopterin (BH4) as a cofactor, converts phenylalanine (Phe) into tyrosine. If left untreated, the accumulation of Phe leads to irreversible damage in the brain, resulting in global developmental delay, intellectual disability, and various other neuropsychiatric problems.1
The frequency of elevated Phe (hyperphenylalaninemia, HPA) varies depending on country, region, and ethnicity. The incidence is estimated to be 1:10,000 in Europe2, 1:25.000 in the United States, and 1:15,924 in China.3 In recent studies published worldwide, it is noted that Türkiye has the highest incidence of PKU.4,5 Özalp et al.6 reported, the incidence of PKU in Türkiye as 1:4,172 in 1986. Based on the latest report by the Ministry of Health, the incidence of phenylketonuria in Türkiye is 1 in 4,500 live births.7
In the 1960s, Robert Guthrie developed a microbiological test, leading to the initiation of the first newborn screening (NBS) program for PKU in Massachusetts, USA, in 1963.8 Later, more sensitive methods, such as fluorometric and spectrometric techniques, were developed. Türkiye started a pilot program in 1983, expanding it nationwide in 2006.9 Ideally, newborns with PKU should be diagnosed and start treatment within the first fifteen days of life. Current guidelines classify patients who begin treatment after three months as “late-diagnosed”.2
This study represents the clinical characteristics of 1103 patients identified through an NBS program. By examining demographic data, clinical findings, and diagnostic results, we aimed to provide valuable insights that can enhance the workings of the NBS program.
Materials and Methods
Study design and participants
In this retrospective study, the health records of infants who were referred with suspicion of PKU through the national NBS program to the Division of Pediatric Metabolism at Hacettepe University İhsan Doğramacı Children’s Hospital between January 2016 and January 2023 were reviewed. Patient information was obtained from the parents. The study was conducted in accordance with the ethical principles outlined in the Declaration of Helsinki and was approved by the Hacettepe University Ethics Committee for Non-Interventional Clinical Studies.
Sample collection and screening protocol
According to the protocol of the national NBS program, capillary blood samples collected from the newborns using a Guthrie card at healthcare institutions are sent as dried blood spots (DBS) to designated screening laboratories by the Ministry of Health. For samples with a Phe level of 2.1 mg/dL or higher, additional diagnostic and clinical follow-up actions are initiated. The blood test results, conducted by the screening laboratory and transmitted to the provinces through the NBS Program Web Application, lead to infants suspected of having PKU being referred to pediatric nutrition and metabolism clinics by their registered family physicians. The Ministry’s screening algorithm does not include a third DBS; however, the results of patients who had a third DBS collected were also documented (Fig. 1), which may have been collected due to previous samples being improperly collected or shipped. The “time of DBS result” was defined as the interval between the sample collection date and the result finalization date. The “age at admission “ was defined as the age of the patient (in days) when they first applied to our center. “Late admission” was defined as age at admission older than 90 days.
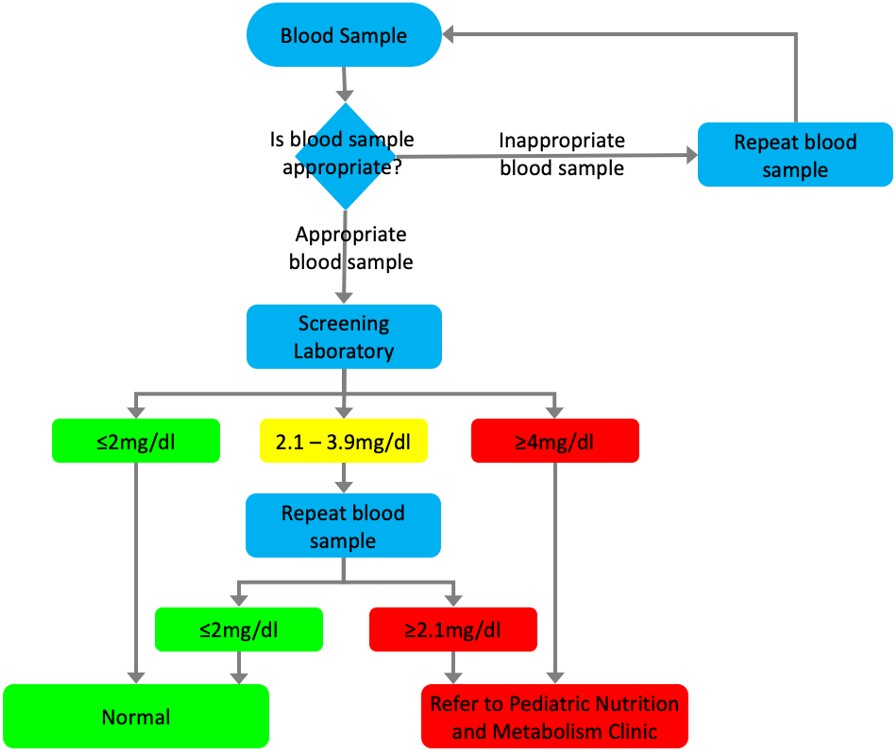
There are various classifications for PKU.2,10-12 We classified patients as follows: patients who do not require treatment (Group 1; untreated Phe level 2–6 mg/dL), and patients who require treatment (Group 2; untreated Phe level 6–20 mg/dL, and Group 3; untreated Phe level >20 mg/dL). Transient HPA and transient tyrosinemia are temporary conditions, characterized by elevated blood phenylalanine (≥2.1 mg/dL) or tyrosine (>3 mg/dL) levels, usually detected during routine NBS, normalizing spontaneously at follow-up. In our center, phenylalanine and tyrosine levels were measured using quantitative amino acid analysis by high performance liquid chromatography.
Statistical analyses
Data analysis was performed using SPSS v26.0 (SPSS Inc., Chicago, USA). Distribution of the variables was assessed using both visual (histograms, probability plots) and analytical methods (Kolmogorov-Smirnov and Shapiro-Wilk tests). Descriptive statistics were reported as mean ± standard deviation (SD) for normally distributing quantitative data, as median, range (minimum–maximum), and interquartile range (IQR, Q1-Q3) for non-normally distributing quantitative data, and as frequencies and percentages for qualitative data. The Mann-Whitney U test was utilized to assess the differences between two independent groups with non-parametric data. Depending on the test conditions, either chi-square or Fisher’s exact tests were employed to compare categorical variables. As DBS test features, age at admission, plasma Phe level were not normally distributed the Kruskal- Wallis tests were conducted to compare these parameters and the ordinal variables among the geographical regions (7 groups). The Mann-Whitney U test was performed to test the significance of pairwise differences using Bonferroni correction (p=0.0023) to adjust for multiple comparisons when investigating the associations between non-normally disturbed variables. The correlation coefficients and their significance were calculated using the Spearman test. The univariate analyses to identify variables associated with age at admission were investigated using Mann-Whitney U test. In univariate analyses, variables were selected for inclusion in multivariate analysis based on statistically significant factors, typically with a p-value <0.05. In addition, parameters considered to be clinically relevant or potentially influential were included. Thus, the following variables were included in the model: family history of PKU, geographical region, and 1st DBS result. For the multivariate analysis, the possible factors identified with univariate analyses were further entered into the logistic regression analysis to determine independent predictors of patient “age at admission” and “late admission”. Logistic regression model was constructed by enter method, using variables associated with “age at admission” and “late admission”. Hosmer-Lemeshow goodness of fit statistics were used to assess the fitness of the logistic regression model. To preserve the integrity of the model in the presence of multicollinearity, only one variable from each highly correlated set was incorporated. The cut-off value of the first DBS result in predicting the age at admission was analyzed using receiver operating characteristics (ROC) curve analysis. While evaluating the area under the curve, a 5% type-I error level was used to accept a statistically significant predictive value of the test variables.
Results
Overview of patient characteristics
1103 patients referred from the national NBS program for elevated Phe level between January 2016 and January 2023 were included. The demographic and neonatal clinical characteristics of the patients, and the initiated treatments, and the final diagnoses are shown in Table I, and the laboratory findings are shown in Table II. The majority of referred infants were either normal (38.1%) or had mild HPA not requiring treatment (38.3%), and the rest had transient or hereditary disorders of phenylalanine or tyrosine metabolism, or a variety of diagnoses unrelated to this pathway, most commonly galactosemia. In total, there were 140 infants who required treatment.
|
*Others: Arrhythmia, early rupture of membranes, hypoglycemia, hypoxic-ischemic encephalopathy, necrotizing enterocolitis, intrauterine growth restriction, polycythemia, respiratory distress syndrome ** Group 1: untreated Phe level 2–6 mg/dL (patients not requiring treatment); Group 2: untreated Phe level 6–20 mg/dL (patients requiring treatment); Group 3: untreated Phe level >20 mg/dL (patients requiring treatment). HPA, hyperphenylalaninemia; Phe, phenylalanine; PKU, phenylketonuria; Tyr, tyrosine. |
|
| Table I. General characteristics of infants referred through newborn screening for phenylketonuria (N=1103). | |
| Characteristics | |
| Female sex, n (%) |
|
| Consanguinity, n (%) |
|
| Maternal age, yr, mean ± SD |
|
| Paternal age, yr, mean ± SD |
|
| Family history of PKU, n (%) |
|
| Gestational age, wk, mean ± SD |
|
| Birth weight, gr, mean ± SD |
|
| History of neonatal hospitalization, n (%) |
|
| Neonatal jaundice |
|
| Neonatal respiratory distress |
|
| Premature birth |
|
| Congenital pneumonia |
|
| Urinary tract infection |
|
| Sepsis |
|
| Others* |
|
| History of neonatal jaundice, n (%) |
|
| Phototherapy, n (% among jaundice) |
|
| No treatment, n (% among jaundice) |
|
| Age at admission, days, median (Q1-Q3), min-max |
|
| 1st plasma Phe level (at our center), mg/dL (n=1103), median (Q1-Q3), min-max |
|
| 1st plasma Tyr level (at our center), mg/dL (n=1103), median (Q1-Q3), min-max |
|
| Diagnosis, n (%) | |
| Normal |
|
| Disorders of Phe or Tyr metabolism | |
| Group 1; untreated Phe level 2–6 mg/dL |
|
| Group 2 PKU; untreated Phe level 6–20 mg/dL |
|
| Group 3 PKU; untreated Phe level >20 mg/dL |
|
| Defects of BH4 metabolism or DNAJC12 |
|
| Transient tyrosinemia |
|
| Transient HPA |
|
| HPA + transient tyrosinemia |
|
| Transient HPA + transient tyrosinemia |
|
| Transient tyrosinemia + Group 2 PKU |
|
| Maternal PKU syndrome |
|
| Other inherited metabolic disorders |
|
| Galactosemia |
|
| Citrin deficiency |
|
| I-cell disease |
|
| Lipoprotein lipase deficiency |
|
| Follow up / Treatment, n (%) | |
| No follow-up required |
|
| Follow-up required |
|
| Lost to follow-up |
|
| Followed without treatment |
|
| Followed with treatment |
|
| Diet |
|
| Sapropterin monotherapy for PKU |
|
| Neurotransmitter precursors and sapropterin |
|
|
Values given as median (Q1-Q3), min-max, followed by the number of infants (n). DBS, dried blood spot, Phe, phenylalanine. |
|||
| Table II. DBS timing and phenylalanine levels in infants referred via newborn screening (N=1103). | |||
|
|
|
|
|
| Age at collection (days) |
|
|
|
| Time it took for the result (days) |
|
|
|
| Phe level (mg/dL) |
|
|
|
The timing of DBS collections and DBS results within the NBS program are summarized in Table II. 26 infants (2.36%) had late admissions (after 90 days). 18 of these infants had normal Phe levels, 5 had HPA, 2 had PKU with untreated Phe level >20 mg/dL, and 1 had transient tyrosinemia.
Comparison of geographical regions
Significant differences were observed among the geographical regions in Türkiye, regarding the age at the 1st, 2nd, and 3rd DBS samples, and the time it took for the 1st and 2nd DBS tests to finalize as well as the age at admission to the center. In the Black Sea region, the time for sample collection and result finalization was later compared to the Central Anatolia region. In the Southeastern and Eastern Anatolia regions, the collection time for the 3rd DBS, the time of 1st DBS result, and the age at admission were later compared to the Central Anatolia region. In the Black Sea region, the age of 1st DBS collection was later when compared to the Southeastern and Eastern Anatolia regions, but the age at admission to our center was found to be earlier. In the Aegean region, the time of 1st DBS result and time of 2nd DBS result was earlier when compared to the Eastern Anatolia region. Patients from the Central Anatolia and Marmara regions were younger at the age of admission (Supplementary Table S1 and S2).
Comparison of clinical and laboratory characteristics across patient groups
The median age at admission was 20.5 days (Q1-Q3: 17.5-25.5, min-max: 12-62 days) for patients with first DBS Phe levels higher than 20 mg/dL (n=12), and 22.5 days (Q1-Q3: 13-36.5, min-max:12-42 days) for patients with second DBS results higher than 20 mg/dL (n=4).
Supplementary Table S3 shows differences in the 1st, 2nd, and 3rd DBS results and the initial plasma Phe levels among groups categorized by neonatal hospitalization status, low birth weight status, history of antibiotic use, preterm and term births, presence of neonatal jaundice, and phototherapy treatment. No significant differences in Phe levels were observed between the Transient HPA (n=26) and Transient Tyrosinemia (n=56) groups regarding neonatal hospitalization or antibiotic use (p=1, and p=0.86, respectively). However, a history of neonatal jaundice was 4.5 times more common in the Transient Tyrosinemia group compared to the Transient HPA group, and this difference was statistically significant (p=0.04). No significant differences were observed in gestational age, birth weight, 1st DBS, 2nd DBS, and 3rd DBS results (Supplementary Table S3).
There were no significant differences in gestational age and birth weight parameters between the HPA (n=422) and normal (n=422) groups (p=0.07-0.655). However, there were significant differences in the 1st, 2nd, and 3rd DBS Phe levels (p<0.001) between normal and HPA groups. The DBS Phe levels were higher in the HPA group (Supplementary Table S3).
Patients with transient HPA, transient tyrosinemia, and transient HPA combined with transient tyrosinemia were recorded as a single “transient” group and compared with the normal group (Supplementary Table S3). There were no differences in gestational age, birth weight, 2nd DBS, and 3rd DBS values. The risk of neonatal hospitalization in the transient group was 1.6 times higher compared to the normal group (p=0.04). The history of jaundice in the transient group was 2.7 times more common compared to the normal group (p=0.001).
Determinants of age at admission
There was a statistically significant relationship between having a family history of PKU and age at admission: The median day of admission was 19 (3-86) days for patients with a family history of PKU and 26 (4-410) days for those without (p<0.001). For patients with Phe levels measured at our center below 4 mg/dL (n=504), the median age at admission was 26 days (Q1-Q3: 21-34, min-max: 4-410), while for patients with Phe levels above 4 mg/dL (n=209), the median age at admission was 14 days (Q1-Q3: 10-20, min-max: 4-138), and this difference was statistically significant (p<0.001). Spearman correlation analysis between the 1st DBS Phe level and age at admission to our center showed a moderate, negative correlation (r= -0.41, p<0.001). ROC curve analysis for predicting early admission based on the 1st DBS Phe level yielded a cut-off value of 4.05 mg/dL (area under the curve: 0.816, 95% confidence interval: 0.777–0.854, p<0.001, sensitivity: 90.2%, specificity: 68%; Supplementary Fig. S1). Logistic regression analysis showed that positive family history of PKU, certain geographical regions, higher 1st DBS Phe level were associated with earlier age at admission (Table III). In Eastern Anatolia, Southeastern Anatolia, Mediterranean and the Black Sea regions, the age of admission was found to be significantly later (Supplementary Table S1 and S2).
|
The model is statistically significant with p <0.001; Hosmer- Lemeshow: 0.446; Nagelkerke R2: 0.442; overall accuracy: 84.9%. In the second logistic regression model examining factors influencing late admission, the region was found to be a significant factor with p=0.002; Hosmer-Lemeshow: 0.233; Nagelkerke R2: 0.050; overall accuracy: 97.8%. All regions were included in the model separately. Descriptive and comparative characteristics of the differences between regions are given in Supplementary Table S1 and S2. CI, confidence interval; DBS, dried blood spot; Phe, phenylalanine; PKU, phenylketonuria. |
|||||
| Table III. Significant predictors of age at admission and late admission by logistic regression. | |||||
| Variables in model |
|
|
|
|
|
| Significant predictors of “age at admission” | |||||
| Constant |
|
|
|
|
|
| Family History of PKU |
|
|
|
|
|
| Region |
|
|
|
|
|
| 1st DBS Phe level |
|
|
|
|
|
| Significant predictors of “late admission” | |||||
| Constant |
|
|
|
|
|
| Region |
|
|
|
|
|
Discussion
The results of our study highlight several critical findings related to the clinical characteristics of patients identified through the national NBS program for disorders of Phe metabolism, and to the workings of the NBS program itself. Our comprehensive analysis of 1103 patients offers valuable insights into the demographics, clinical features, and final diagnosis.
Regional disparities in newborn screening and healthcare access
One of the significant findings of our study is significant geographical differences in the timing of DBS sample collection, result reporting and admission time, which could reflect variations in healthcare infrastructure, accessibility, and geographical challenges. These disparities can be attributed to factors such as healthcare infrastructure, availability of adequate healthcare workers, and health policies. Additionally, family-related factors such as beliefs, education level, and awareness may contribute to the delays in collecting and transporting DBS samples to the center. Efforts should be made to speed up sample transport and analysis, either by establishing a new screening center closer to these regions or by optimizing logistical algorithms for more efficient sample processing and transfer.
Detection of transient and secondary metabolic conditions
Transient tyrosinemia and transient HPA result from the immaturity of hydroxylation enzymes or hepatocyte damage. Transient tyrosinemia is also associated with ascorbic acid deficiency and prematurity.13 Transient conditions can also occur in other metabolic diseases causing liver damage, such as galactosemia and citrin deficiency. The incidence of transient tyrosinemia of the newborn varies between 0.2-10%.14 We observed an incidence of 5.1% among the referred infants, consistent with existing data. Studies have shown that maternal/neonatal complications are more frequent in the Transient Tyrosinemia group, but long-term cognitive outcomes do not differ from those of normal infants.15,16 We demonstrated that jaundice was more prevalent in the Transient Tyrosinemia group compared to the Transient HPA group. Furthermore, the comparison of Transient HPA and/or Transient Tyrosinemia with normal patients showed higher rates of neonatal jaundice and hospitalization in the transient group. To our knowledge, no previous studies have compared these groups. Our results suggest that mild Phe or tyrosine elevations detected by NBS are more likely to be transient if the newborn has a history of jaundice or hospitalization, which can guide counseling.
In our study, we identified patients with various other inherited metabolic disorders, diagnosed incidentally. These included galactosemia (n=7), citrin deficiency (n=1), I-cell disease (n=1) and lipoprotein lipase deficiency (n=1). Studies from our country have reported cases of citrin deficiency, as documented by Zeybek et al.17; while Yekeduz et al.18 and Unal et al.19 reported cases of maple syrup urine disease, galactosemia, and tyrosinemia. According to the literature, Shakespeare et al.20 reported cases of galactosemia (n=8), biopterin defects (n=2), and tyrosinemia type 1 (n=1). In our study, we identified patients with DNAJC12 deficiency (n=1) and disorders of BH4 metabolism, which include 6-pyruvoyltetrahydropterin synthase deficiency (n=3) and dihydropteridine reductase deficiency (n=1), which are among the secondary aims of NBS for PKU. However, there have been no reported cases of I-cell disease, and lipoprotein lipase deficiency. The patient is the first to be diagnosed with lipoprotein lipase deficiency based on findings from the NBS, where inaccurately elevated measurement of the patient’s blood Phe levels due to the lipemic nature of the sample ultimately led to the diagnosis. Additionally, one patient was diagnosed with congenital leukemia presenting with massive hepatomegaly. This patient has been reported previously.21 Careful physical examination to detect signs such as cholestasis, dysmorphism, leukocoria, neurological findings, and hepatomegaly is crucial, especially in patients identified through screening, as it helps identify other disorders, including other inherited metabolic disorders.
We identified five cases of maternal PKU syndrome presenting with severe congenital anomalies, consistent with previous studies.22,23 Timely DBS collection is crucial, as delayed sampling may lead to the normalization of the newborn’s Phe levels, potentially missing the diagnosis of maternal PKU. NBS programs offer secondary benefits, including the detection of maternal metabolic disorders.10,24,25
Impact of family history and referral threshold on early diagnosis
Our study revealed that there is substantial variation in the age at admission based on a family history of PKU. Patients with a positive family history presented earlier compared to those without such a history. It is essential to consider that these families might seek medical attention proactively without waiting for a referral. In line with the Ministry algorithm, patients with an initial Phe level above 4 mg/dL are referred immediately without waiting for a second sample. Our analysis confirms that a first DBS Phe level above 4.05 mg/dL is associated with an earlier clinical presentation. This finding aligns with the Ministry algorithm, suggesting that the current referral threshold effectively facilitates earlier medical consultation and diagnosis.
Variability in false positive rates due to screening methods and cutoff level variations
In our study, 38% of individuals with elevated phenylalanine levels in NBS were determined to have Phe within normal limits. The false positive rate for PKU screening in Türkiye ranges from 44% to 76.2%.18,26,27 Studies conducted in different regions of the world have reported false positive rates ranging from 81% to 94%.4,28 While direct comparisons are complicated by disparities in measurement techniques and threshold values, the false positive rate identified at our center seems to reside within an acceptable spectrum. Systematic annual dissemination of data regarding false positive and false negative rates, in addition to the positive and negative predictive values by the Ministry of Health, would be instrumental in enhancing and refining the newborn screening initiative.
Considerations for optimizing the Turkish PKU NBS Program
Considering our findings, it is imperative to enhance the NBS programs and follow-up protocols. The early detection and treatment of PKU is crucial in preventing severe long-term complications. Based on our findings, several measures can be taken to prevent delayed diagnoses, both across the entire population and in specific regions. Firstly, increasing the accessibility and efficiency of healthcare services in regions with identified delays, such as Eastern Anatolia, Southeastern Anatolia, Mediterranean and the Black Sea regions, is crucial. This can be achieved by improving healthcare infrastructure, ensuring the availability of adequate healthcare workers, and implementing targeted health policies. It is necessary to educate families about the importance of timely screening and follow-up, particularly in regions with lower levels of awareness.
This study is one of the largest single-center studies conducted examining NBS. It offers a comprehensive analysis of demographic data, clinical findings, and diagnostic results, contributing valuable insights into the workflow of NBS programs. The study highlights regional disparities in healthcare access, guiding policy decisions to improve healthcare delivery in underserved areas. The occurrence of two delayed diagnoses in patients with a classical PKU is regrettable, and similar situations are likely to have occurred in other centers. To prevent delayed diagnoses, it is crucial to implement strategies that ensure timely and accurate screening and follow-up.
Our study has some limitations. The study’s retrospective design relies on the accuracy and completeness of historical medical records, which may introduce biases or data inaccuracies. As a single-center study, the findings may not be generalizable to other regions or countries with different healthcare infrastructures and practices. Given that the national NBS program is centrally managed by the Ministry of Health, open access to screening processes and outcome data would facilitate further improvements. Transparent data sharing could help refine screening protocols, address regional disparities and optimize the overall effectiveness of the NBS
In conclusion, our study underscores the importance of timely and accurate NBS for phenylalanine metabolism disorders. By improving early diagnostic and follow-up practices, we can significantly impact the clinical outcomes and quality of life of affected individuals. Future research should focus on addressing regional disparities and optimizing NBS protocols to ensure that all newborns receive the best possible start in life.
Ethical approval
The study was approved by Hacettepe University Ethics Committee (date: 21.09.2021, number: 15/44, 2021/09).
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet 2010; 376: 1417-1427. https://doi.org/10.1016/S0140-6736(10)60961-0
- van Wegberg A, MacDonald A, Ahring K, et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis 2017; 12: 162. https://doi.org/10.1186/s13023-017-0685-2
- Hillert A, Anikster Y, Belanger-Quintana A, et al. The genetic landscape and epidemiology of phenylketonuria. Am J Hum Genet 2020; 107: 234-250. https://doi.org/10.1016/j.ajhg.2020.06.006
- Lin Y, Lin W, Su R, Zheng Z, Fu Q, Wang G. Newborn screening and genetic features of patients with hyperphenylalaninemia in a southern Chinese population. Clin Chim Acta 2022; 535: 13-18. https://doi.org/10.1016/j.cca.2022.08.009
- Shoraka HR, Haghdoost AA, Baneshi MR, Bagherinezhad Z, Zolala F. Global prevalence of classic phenylketonuria based on Neonatal Screening Program Data: systematic review and meta-analysis. Clin Exp Pediatr 2020; 63: 34-43. https://doi.org/10.3345/kjp.2019.00465
- Ozalp I, Coşkun T, Tokatli A, et al. Newborn PKU screening in Turkey: at present and organization for future. Turk J Pediatr 2001; 43: 97-101.
- Newborn Metabolic and Endocrine Disease Screening Program (NTP). Available at: https://hsgm.saglik.gov.tr/tr/tarama-programlari/ntp.html (Accessed December 4, 2024).
- Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 1963; 32: 338-343.
- Arısoy A, Arısoy E, Ayvaz A, Coşkun T, Özalp İ. Van ili’nde 3117 yenidoğanda fenilketonüri taraması. Çocuk Sağlığı ve Hastalıkları Dergisi 1992; 35: 265-269.
- Coşkun T, Çoker M, Mungan NÖ, Özel HG, Sivri HS. Recommendations on phenylketonuria in Turkey. Turk J Pediatr 2022; 64: 413-434. https://doi.org/10.24953/turkjped.2021.4098
- Camp KM, Parisi MA, Acosta PB, et al. Phenylketonuria Scientific Review Conference: state of the science and future research needs. Mol Genet Metab 2014; 112: 87-122. https://doi.org/10.1016/j.ymgme.2014.02.013
- Vockley J, Andersson HC, Antshel KM, et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet Med 2014; 16: 188-200. https://doi.org/10.1038/gim.2013.157
- Zea-Rey AV, Cruz-Camino H, Vazquez-Cantu DL, Gutiérrez-García VM, Santos-Guzmán J, Cantú-Reyna C. The incidence of transient neonatal tyrosinemia within a Mexican population. Journal of Inborn Errors of Metabolism and Screening 2017; 5: 2326409817744230. https://doi.org/10.1177/2326409817744230
- WHO Scientific Group. Screening for inborn errors of metabolism: report of a WHO scientific group [meeting held in Geneva from 20 to 26 November 1967]. World Health Organ Tech Rep Ser 1968; 401: 1-57.
- Menkes JH, Chernick V, Ringel B. Effect of elevated blood tyrosine on subsequent intellectual development of premature infants. J Pediatr 1966; 69: 583-588. https://doi.org/10.1016/s0022-3476(66)80044-6
- Light IJ, Sutherland JM, Berry HK. Clinical significance of tyrosinemia of prematurity. Am J Dis Child 1973; 125: 243-247. https://doi.org/10.1001/archpedi.1973.04160020067013
- Zeybek AC, Kiykim E, Zubarioglu T, Cansever MS, Ceylaner S, Erkan T. Citrin deficiency: an infant incidentally detected by phenylketonuria screening with a novel mutation in slc25a13 gene. Genet Couns 2015; 26: 409-413.
- Koç Yekedüz M, Eminoğlu FT. Diagnosis and treatment of newborns referred to the metabolism department from the national newborn screening program in Türkiye: a 5-year single-center experience. Turkish Journal of Pediatric Disease 2024; 18: 274-280. https://doi.org/10.12956/tchd.1454353
- Unal O, Oztürk-Hişmi B, Coşkun T, Tokatlı A, Dursun A, Sivri HS. Detection of other inborn errors of metabolism in hyperphenylalaninemic babies picked up on narrow-spectrum screening programs. Turk J Pediatr 2012; 54: 409-412.
- Shakespeare L, Downing M, Allen J, et al. Elevated phenylalanine on newborn screening: follow-up testing may reveal undiagnosed galactosaemia. Ann Clin Biochem 2010; 47: 567-569. https://doi.org/10.1258/acb.2010.010083
- Yaman Bajin İY, Yıldız Y, Akın Ş, et al. Infant acute lymphoblastic leukemia with atypical presentation. Acta Medica 2019; 50: 57-59. https://doi.org/10.32552/2019.ActaMedica.406
- Yıldız Y, Sivri HS. Maternal phenylketonuria in Turkey: outcomes of 71 pregnancies and issues in management. Eur J Pediatr 2019; 178: 1005-1011. https://doi.org/10.1007/s00431-019-03387-8
- Savli P, Ersoy M, Guner AE, Tas I. Evaluation of babies with hyperphenylalaninemia diagnosed in the National Newborn Screening Program in Istanbul in 2019. International Journal of Medical Biochemistry 2022; 5: 49-53. https://doi.org/10.14744/ijmb.2021.44127
- Tin O, Zübarioğlu T, Cansever MŞ, Kıykım E, Aktuğlu-Zeybek Ç. Maternal inborn errors of metabolism detected in expanded newborn metabolic screening. Turk Arch Pediatr 2023; 58: 382-387. https://doi.org/10.5152/TurkArchPediatr.2023.23009
- Schimmenti LA, Crombez EA, Schwahn BC, et al. Expanded newborn screening identifies maternal primary carnitine deficiency. Mol Genet Metab 2007; 90: 441-445. https://doi.org/10.1016/j.ymgme.2006.10.003
- Toktaş İ, Sarıbaş S, Canpolat S, Erdem Ö, Özbek MN. Evaluation of patients diagnosed with phenylketonuria and biotinidase deficiency by the newborn screening program: a ten-year retrospective study. Turk J Pediatr 2022; 64: 985-992. https://doi.org/10.24953/turkjped.2022.467
- Kazanasmaz H, Karaca M. Comparison of fluorometric immunoassay and tandem mass spectrometry methods used in the diagnosis of phenylalanine metabolism disorders in Turkish children. Istanbul Medical Journal 2019; 20: 279. https://doi.org/10.4274/imj.galenos.2019.53189
- Abbaskhanian A, Zamanfar D, Afshar P, et al. Incidence of neonatal hyperphenylalaninemia based on high-performance liquid chromatography confirmatory technique in Mazandaran province, Northern Iran (2007-2015). Int J Prev Med 2017; 8: 93. https://doi.org/10.4103/ijpvm.IJPVM_24_17
Copyright and license
Copyright © 2025 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









