
Abstract
Background. Osteogenesis imperfecta (OI) is a genetic disorder of connective tissues caused by an abnormality in the synthesis or processing of type I collagen. The combination of OI and inflammatory arthritis is rare. Our literature review identified 5 cases of OI-related inflammatory arthritis, but only 2 of these cases have been reported in children.
Case Report. We present 3 cases diagnosed with OI and juvenile idiopathic arthritis (JIA). Two were diagnosed with enthesitis-associated arthritis, and one was diagnosed with oligoarticular JIA with laboratory findings and a magnetic resonance imaging examination. Only one of the patients had a previously diagnosed OI. For the others, whole gene sequence analysis was performed, and a mutation in the collagen type I alpha 1 (COL1A1) gene was detected. Identifying and treating inflammatory arthritis in our patients with OI improved their joint pain.
Conclusion. Musculoskeletal pain is a common issue in individuals with OI and JIA. Considering children with OI may also develop arthritis, early diagnosis, and accurate treatment may be crucial. Recognizing the rare association between JIA and OI is important, as investigating this relationship could help alleviate the disease burden. Thorough evaluation and prompt diagnosis of JIA in patients with OI can significantly reduce the impact of the disease.
Keywords: osteogenesis imperfecta, juvenile idiopathic arthritis, arthralgia, bone mineral density, recurrent fractures
Introduction
Juvenile idiopathic arthritis (JIA) is the most common rheumatologic disease in childhood. The incidence and prevalence reported in European and North American populations ranged from 2 to 20 and 16 to 150 per 100,000, respectively.1 A multifactorial interplay between immune, genetic, and environmental factors shapes the etiopathogenesis of JIA. The primary characteristic of JIA is tissue destruction with joint inflammation.2 The synovium becomes thickened due to the uncontrolled proliferation of various cells, including synoviocytes and immune cells such as B cells, T cells, natural killer cells, macrophages, neutrophils, plasma cells, and dendritic cells. These immune cells infiltrate the sub-lining layer of the synovium. This immune response not only contributes to ongoing joint inflammation but also causes long-term structural damage, including cartilage degradation and bone erosion.3 Disease classification is determined according to the criteria set by the International League of Associations for Rheumatology (ILAR) and is divided into seven subtypes.1
Osteogenesis imperfecta (OI), also known as “brittle bone disease,” is a rare inherited connective tissue disorder. The estimated incidence is one in 10,000 to 20,000 births. It is often a disease caused by pathogenic variants in either collagen type I alpha 1 (COL1A1) or collagen type I alpha 2 (COL1A2), which encode components of type I collagen. Recent studies have identified defects in multiple genes that encode proteins involved in the synthesis, secretion, processing, and post-translational modification of type I collagen. Additionally, mutations in proteins that regulate the differentiation and activity of bone-forming cells have been shown to cause OI. For instance, in cases of type I collagen mutations or mutations that affect the proteins involved in its biosynthetic pathway, osteoblasts frequently exhibit enlarged endoplasmic reticulum (ER) cisternae and signs of ER stress. This leads to a decreased secretion of type I collagen into the extracellular matrix, increased mineralization of the matrix, and impaired communication between the matrix and cells. This results in less than normal bone trabeculation, thinner bones and therefore brittle bones.4 The clinical phenotypes of OI vary from perinatal death to mild forms without fractures. The revised Nosology and Classification of Genetic Skeletal Disorders defines five clinical forms of OI: non-deforming with persistently blue sclera (OI type I), perinatal lethal (OI type II), progressively deforming (OI type III), moderate (OI type IV), and with calcification of the interosseous membranes and/or hypertrophic callus (OI type V). However, patients have some common clinical features, such as an increased risk of fractures, skeletal fragility, and varying degrees of bone deformities.
The disease can affect any tissues that have type I collagen.5 Joint findings such as arthralgia and deformities due to fractures may be missed if there is an accompanying diagnosis of JIA.
JIA and OI are distinct diseases, each with heterogeneous clinical manifestations. The concurrence of OI and JIA in a patient is extremely rare due to several factors, including the differences in their underlying pathophysiological mechanisms, genetic causes, and specific clinical features, which typically do not overlap.6 OI involves structural bone abnormalities resulting from mutations in the collagen synthesis pathway, whereas JIA is primarily characterized by autoimmune-induced joint inflammation. While JIA can result in long-term bone damage, primarily through mechanisms of joint inflammation and bone erosion, its primary pathological process is synovitis, which is different from the bone-related issues associated with OI. The absence of significant inflammatory processes in OI, combined with the absence of common genetic pathways, further reduces the likelihood of these two diseases occurring simultaneously. So far, two cases of polyarticular JIA and three cases of seropositive rheumatoid arthritis (RA) have been reported with OI.6-9 We report three cases of JIA associated with OI and review the literature.
Case Presentations
Case 1
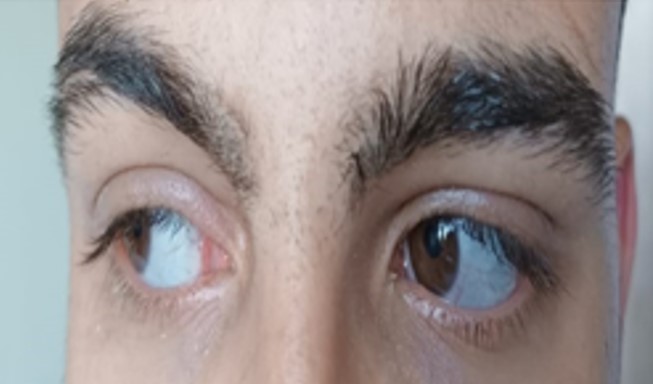
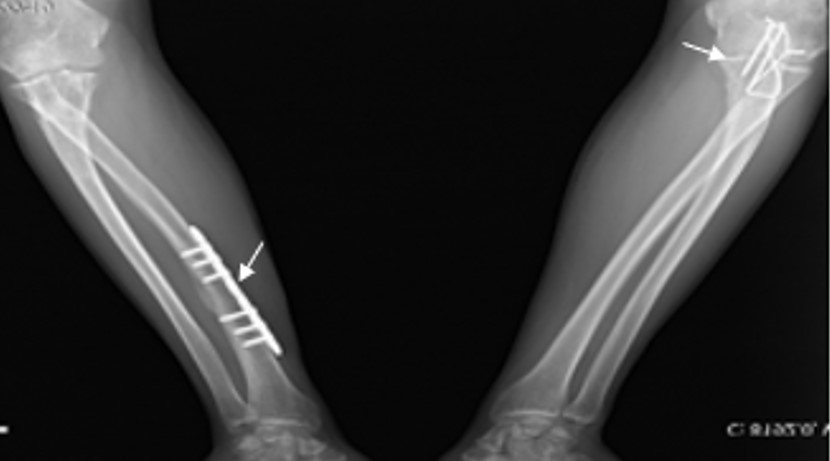
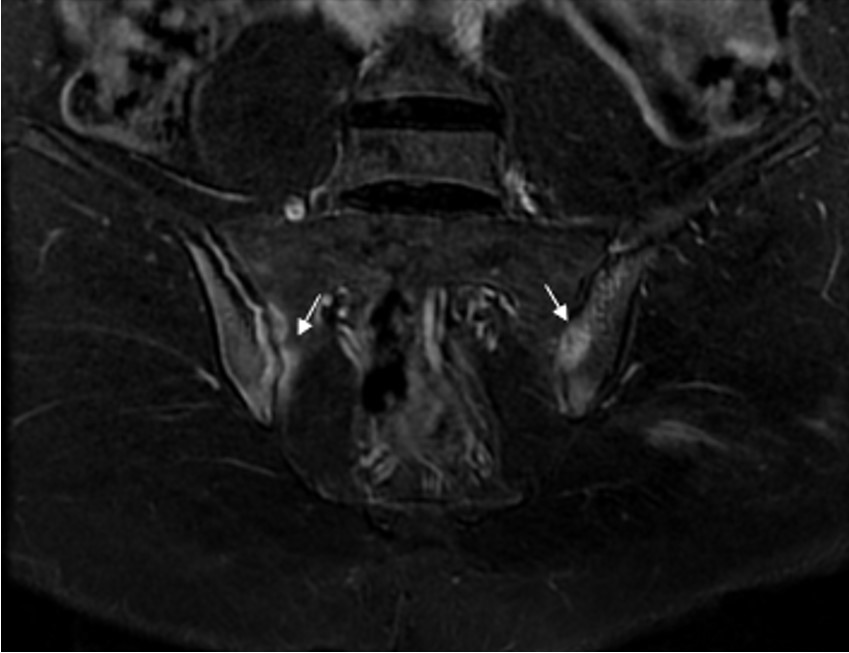
A 17-year-old boy was admitted to our Pediatric Rheumatology Clinic due to hip joint pain and morning stiffness persisting for three years. His medical history revealed seven recurrent fractures of the upper extremities, three of which required surgery. According to the family pedigree, many family members had also experienced recurrent fractures (Fig. 1). During the physical examination, the patient’s height was measured at 176 cm (55th percentile) and his weight at 66 kg (35th percentile). The patient exhibited blue sclera, pectus excavatum, restricted motion in the right hip joint, sacroiliac joint pain, positive bilateral flexion abduction external rotation (FABER) and Schober test results, as well as surgical scar marks on his arms (Fig. 2). The brain stem evoked response auditory (BERA) test revealed mild sensorineural hearing loss. Laboratory parameters included white blood cell (WBC): 7,130/mm3, hemoglobin (Hb): 13.7 g/dL, platelet (PLT): 223,000/mm3, erythrocyte sedimentation rate (ESR) 7 mm/hour, and C-reactive protein (CRP): 7.1 mg/L (average: 5 mg/L). He had normal vitamin D, alkaline phosphatase (ALP), parathyroid hormone, phosphorus, and calcium levels. Human leukocyte antigen-B27 (HLA-B27) was positive. Rheumatoid factor (RF) and antinuclear antibodies (ANA) were negative. X-rays of the hip and radius showed a significant decrease in bone density and trabecular prominence (Fig. 3). Bone mineral density (BMD) revealed the Z-score of the lumbar spine (L1-L4) was -3. The patient was diagnosed with OI after a heterozygous pathogenic variant (NM_000088.3) was detected in the COL1A1 gene c.3749del (p.Gly1250Alafs*81) by whole exome sequencing. Intravenous zoledronate treatment was given every six months. Magnetic resonance imaging (MRI) revealed bilaterally chronic sacroiliitis (Fig. 4). Methotrexate treatment was started with the diagnosis of enthesitis-related arthritis (ERA), and adalimumab was added three months later due to inadequate improvement in symptoms. In the 6th month of rheumatological treatment, the patient’s hip pain and morning stiffness subsided, and he continued his daily life easily.

Case 2
A 6-year-old boy was admitted to the Pediatric Rheumatology Clinic with complaints of swelling, pain, and morning stiffness in his right ankle, persisting for one month. His medical history included a history of fractures in the right elbow, the fifth finger of the right hand, and vertebrae at different times. On physical examination, his weight was 18 kg (15th percentile), and her height was 108 cm (3rd percentile). He had blue sclera, swelling, and pain in the right ankle and right knee, and increased heat. His hearing function was normal. Laboratory values included WBC: 10,300/mm3, Hb: 10.3 g/dL, PLT: 878,000/mm3, ESR: 66 mm/hour, CRP: 38 mg/L, ALP: 234 U/L, normal parathyroid hormone, calcium and phosphorus levels, and negative ANA. The BMD Z-score of the lumbar spine (L1-L4) was -2.53, and a genetic examination was requested with a preliminary diagnosis of osteogenesis imperfecta. MRI showed dense effusion in the right ankle joint space and tendon sheaths. After other possible causes were ruled out, methotrexate treatment was started with the diagnosis of oligoarticular JIA. Genetic analysis identified a pathogenic, frameshifting, heterozygous novel c.3571delCinsTTCGA (chr17:48264244) mutation in the 48th exon of the COL1A1 gene, leading to a diagnosis of OI. Intravenous pamidronate treatment was given every three months. In the first year of follow-up, re-swelling and pain developed in the right knee and right ankle. Ultrasonography revealed effusion. Sulfasalazine was added to the treatment, and an intra-articular corticosteroid injection was administered to the knee. Methotrexate dose was increased to 20 mg/m2/week subcutaneously and sulfasalazine to 1500 mg/day oral dose. Etanercept was added to the patient’s treatment, whose arthritis persisted, and inflammation markers remained high in the second year of follow-up. The disease became inactive with etanercept treatment. One year later, he had arthritis in his right ankle. MRI showed effusion, tenosynovitis, and bone marrow edema in the navicular bone. Etanercept was switched to adalimumab. In the fourth year of follow-up, when active arthritis in the right ankle recurred, adalimumab and sulfasalazine treatments were stopped, and tocilizumab was started. Joint pain regressed, and inflammatory markers returned to normal levels.
Case 3
A 7-year-old boy with OI and familial Mediterranean fever (FMF) was admitted to the Pediatric Rheumatology Clinic. He complained of pain and limping in his right hip that had been going on for two years. His medical history included recurrent fractures in the arms, shoulders, and right hip since the age of 1 year. He was treated with pamidronate with the diagnosis of OI. After pamidronate treatment, his bone fracture numbers had decreased. On physical examination, his weight was 21 kg (25th percentile), and height was 117 cm (25th percentile). He had blue sclera, restricted motion in the right hip joint, and sacroiliac joint pain. His hearing function was normal. The laboratory results included WBC: 9,400/mm3, Hb: 12.1 g/dL, PLT: 200,000/mm3, ESR of 44 mm/hour, normal CRP, calcium, phosphorus, negative ANA, RF, and HLA-B27. Genetic analysis identified G>T change in intron 1, at the binding site of the specificity protein (Sp) 1 transcription factor of the COL1A1 gene. The last measured BMD Z-score of the lumbar spine (L1-L4) was -0.6. MRI showed focal bone marrow edema, sclerosis, and erosive changes in the sacroiliac joint surface. He was diagnosed with ERA, and methotrexate was added to the treatment. The hip pain regressed one month later, and inflammatory markers returned to normal. Vertebral X-ray and lumbar MRI performed at the 9th month of treatment were normal. Sacroiliac MRI showed minimal bone marrow edema in the left iliac wing and marked improvement compared to the previous MRI. Methotrexate treatment was given for one year.
A written consent form was obtained from the families for this publication.
Discussion
The association of OI and JIA was initially described in 2013.4 The coexistence of these two conditions causes challenges in diagnosis and treatment because managing OI and JIA requires different approaches. Additionally, subtypes of JIA may differ among individuals with OI. Our literature review identified 5 cases of OI-related inflammatory arthritis, as listed in Table I. Only 2 of these cases have been reported in children. The median age of onset of complaints was 3.5 years for OI, 46 years for arthritis, and a 1/4 male/female ratio. Arthritis can occur at any stage of the OI disease. In 2 of the cases, the diagnosis of OI was made earlier than arthritis; in 2 of the cases, the diagnosis of arthritis was made earlier than in OI; and in 1 of the cases, the diagnosis of OI and arthritis were made at the same time. As seen in these cases, it is important to consider JIA when diagnosing OI patients, even if there is a variable temporal relationship and a long-time interval between the diagnosis of the two diseases. More studies are needed to better understand the causes and treatment of the relationship between these two diseases affecting bones and joints.
| BMD, bone mineral density; CRP, C-reactive protein; ERA, enthesitis-related arthritis; ESR, erythrocyte sedimentation rate; HLA, human leukocyte antigen; JIA, juvenile idiopathic arthritis; NA, Not available; OI, osteogenesis imperfecta; RA, rheumatoid arthritis; RF, rheumatoid factor; SI, sacroileitis. | ||||||||
| Table I. Review of cases of osteogenesis imperfecta and inflammatory arthritis. | ||||||||
| Authors, years & reference | Gender | Age at diagnosis of OI | Age at diagnosis of JIA/RA | Onset types of arthritis | Joint manifestations | Genetic result | BMD Z-score | Laboratory tests |
| Bica et al. 20136 | F | 53 years | 15 years | Polyarticular JIA | Symmetric polyarthritis of large and small joints and temporomandibular joint involvement | NA | NA | Elevated ESR, RF (+) |
| Damian et al. 20207 | ||||||||
| Case 1 | F | 39 years | 46 years | Seropositive RA | Joint pain and stiffness in lower limbs | COL1A1 heterozygous pathogenic variant, c.3399del, p.Ala1134Profs*105 | NA | Elevated ESR and CRP |
| Case 2 | F | In adolescence | 70 years | Seropositive RA | Joint pain in knees, ankles, and shoulders | COL1A1 heterozygous pathogenic variant, c.3399del, p.Ala1134Profs*105 | NA | Elevated ESR and CRP |
| Emna et al. 20228 | M | 15 years | 8 years | Polyarticular JIA | NA | NA | -4.2 | Normal CRP |
| Mormile et al. 20229 | F | 43 years | 43 years | Seropositive RA | NA | Pathogenic heterozygous missense variant in COL1A1 gene, NM_000088:c.769G>A, p.(Gly257Arg)(rs72645321; HGMD ID: CM960320) | NA | NA |
| Present case 1 | M | 17 years | 17 years | ERA | Limitation of range of motion and joint pain in sacroiliac | Pathogenic heterozygous variant in COL1A1 gene, NM_000088.3, c.3749del, p.Gly1250Alafs*81 | -3 | Normal ESR and CRP, positive HLA-B27 |
| Present case 2 | M | 6 years | 6 years | Oligoarticular JIA | Joint pain and swelling in knee and ankle | Pathogenic heterozygous variant in COL1A1 gene, c.3571delCinsTTCGA (chr17:48264244) | -2.53 | Elevated ESR and CRP |
| Present case 3 | M | 3 years | 7 years | ERA | Limitation of range of motion and joint pain in sacroiliac | G>T mutation of intron 1 at the binding site of the Sp1 transcription factor of the COL1A1 gene | -0.6 | Elevated ESR |
The management of these two conditions requires multidisciplinary and individualized treatment. Management of OI is primarily supportive and symptomatic, accompanied by physical therapy and orthopedic surgery. The main treatment goals for OI include improving bone strength, decreasing pain, reducing the risk of fractures, improving mobility, and preventing long-term complications. Bisphosphonate therapy is the most commonly used bone-directed pharmacologic treatment.3 Bisphosphonates were beneficial in the treatment of our patients, in terms of bone pain relief and fractures. Annual bone densitometry is recommended to monitor the effectiveness of treatment.
In JIA, ongoing joint inflammation restricts the individual’s ability to function in daily life. Treatment starts with non-steroidal anti-inflammatory drugs (NSAIDs), followed by disease-modifying anti-rheumatic drugs (DMARDs), that are commonly methotrexate, and/or corticosteroid injections. Systemic administration of high-dose corticosteroids has a good short-term effect but does not affect long-term disease outcomes. Moreover, long-term administration is associated with severe side effects, including osteoporosis, immunosuppression, growth suppression, and metabolic effects.3 Patients should be evaluated every 3 months during treatment until the treatment goal is reached. Methotrexate is one of the most commonly used DMARDs in children. It may cause osteopenia in pediatric patients with malignancy; however, low-dose methotrexate used for inflammatory diseases does not adversely affect bone mass.10 Biological treatment using etanercept and infliximab in children with JIA leads to a reduction in disease activity. The beneficial effects of treatment with tumor necrosis factor-alpha (TNF-α) antibodies on the skeleton have also been documented. Simonini was the first to demonstrate an increase in bone mass after one year of etanercept treatment in children with JIA. The reduction in bone loss was associated with a therapeutic response and decreased disease activity.11 Etanercept also promotes linear growth in children with JIA.12 In a study conducted with adults, there was an increase in bone formation and a decrease in bone resorption during the 12-month treatment of rheumatoid arthritis patients with TNF-α blockers.13 Therefore, it can be said that biological agents positively affect bone health in OI patients with JIA.
Most cases of OI are caused by mutations in the COL1A1 or COL1A2 genes, which encode collagen type 1. The link between collagen and integrins plays a vital role in RA pathogenesis. Furthermore, endoplasmic reticulum stresses, possibly caused by misfolding or excess proteins, have been described in both RA and OI, which can activate inflammation by driving cells to apoptosis or autoantigen formation.14 These findings may lead us to believe systemic inflammation plays an important role in developing OI and RA. According to laboratory findings in the available literature, inflammatory markers were elevated in 3 (60%) patients but not in 1 (20%) patients. In our patients, inflammatory markers were elevated in 2 patients yet normal in 1 patient.
However, a reciprocal interaction exists between bone remodeling and inflammatory pathways in OI and RA. In the OI mouse model, the pro-inflammatory cytokines TNF-α and interleukin-1α are elevated, splenomegaly and increased osteoclast progenitors in the spleen suggest chronic inflammation.15 Monocytes in OI highly express TNF-α and synthesize the receptor activator of the nuclear factor kappa B (RANK) ligand (RANKL), which is an important factor of bone erosions in RA.16 In addition, It is conceivable that extracellular matrix irregularity due to structural abnormalities and repeated traumas, including fractures, may trigger arthritis in OI, similar to the onset of post-traumatic arthritis.17
Fracture risk increases in both OI and JIA by different mechanisms. Spontaneous fractures occur in OI patients with deterioration of the bone matrix. On the other hand, fractures in JIA may occur due to increased inflammatory cytokines, decreased secondary bone mass, physical inactivity, or osteoporosis caused by corticosteroids.18 JIA is an important differential diagnosis in patients with spontaneous fractures. Patients with OI generally do not have elevated inflammatory markers or autoantibody positivity, and clinical symptoms include blue sclera, arthralgia, joint hypermobility, and tendon rupture.19 OI can lead to deformities in the hands characterized by swan neck deformities and reversible contractures, and this condition can be confused with JIA.20
Patients with OI and JIA may have joint manifestations such as arthralgia and deformities due to fractures caused by OI. This may cause the diagnosis of JIA accompanying OI to be missed. Therefore, we believe it is important to be aware of the coexistence of these two diseases. The presence of morning stiffness and synovitis detected in our cases and the positive response to disease-modifying anti-rheumatic drugs (DMARDs) and biological treatments support the accuracy of our JIA diagnosis.
Since the association of JIA and OI has rarely been reported, we think this association is coincidental. Further studies are needed to understand the relationship between these two diseases better. It should be remembered that joint inflammation may develop in addition to existing bone pathology in patients diagnosed with OI. Evaluating and monitoring this condition is essential to determine the appropriate management and treatment approach. Therefore, in patients diagnosed with OI with joint complaints, a joint MRI examination should be performed in addition to a plain X-ray. In our patients, finding and treating JIA with OI improved joint pain. Because musculoskeletal involvement affects the quality of life in these patients, increasing awareness of the possible relationship between OI and inflammatory arthritis may help improve the quality of life.
Ethical approval
The study was approved by Uludağ University Ethics Committee (date: 07.02.2024, number: 2024-1/26). Informed consent was obtained from the parents of the child.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Giancane G, Consolaro A, Lanni S, Davi S, Schiappapietra B, Ravelli A. Juvenile idiopathic arthritis: diagnosis and treatment. Rheumatol Ther 2016; 3: 187-207. https://doi.org/10.1007/s40744-016-0040-4
- Twilt M, Pradsgaard D, Spannow AH, Horlyck A, Heuck C, Herlin T. Joint cartilage thickness and automated determination of bone age and bone health in juvenile idiopathic arthritis. Pediatr Rheumatol Online J 2017; 15: 63. https://doi.org/10.1186/s12969-017-0194-9
- Zaripova LN, Midgley A, Christmas SE, Beresford MW, Baildam EM, Oldershaw RA. Juvenile idiopathic arthritis: from aetiopathogenesis to therapeutic approaches. Pediatr Rheumatol Online J 2021; 19: 135. https://doi.org/10.1186/s12969-021-00629-8
- Marini JC, Forlino A, Bachinger HP, et al. Osteogenesis imperfecta. Nat Rev Dis Primers 2017; 3: 17052. https://doi.org/10.1038/nrdp.2017.52
- Marom R, Rabenhorst BM, Morello R. Osteogenesis imperfecta: an update on clinical features and therapies. Eur J Endocrinol 2020; 183: R95-R106. https://doi.org/10.1530/EJE-20-0299
- Bica BE, Ruiz DG, Magalhães Pde A, Barcellos MG, de Azevedo MN. Association between juvenile idiopathic arthritis and osteogenesis imperfecta: case report. Rev Bras Reumatol 2013; 53: 535-537. https://doi.org/10.1016/j.rbr.2013.05.002
- Damian LO, Zmarandache CD, Vele P, Albu A, Belizna C, Crăciun A. Osteogenesis imperfecta and rheumatoid arthritis: is there a link? Arch Osteoporos 2020; 15: 40. https://doi.org/10.1007/s11657-020-0681-3
- Emna H, Soumaya B, Sonia R, Samia J, Safa R, Houda A, Hela S, Mohamed E. 29 Juvenile idiopathic arthritis and osteogenesis imperfecta: an exceptional association. Rheumatology (Oxford) 2022; 61(Suppl 2): keac496.025. https://doi.org/10.1093/rheumatology/keac496.025
- Mormile I, Russo R, Andolfo I, de Paulis A, Rossi FW, Rendina D. Rheumatoid arthritis and osteogenesis imperfecta: is there a genetic causal association? Osteoporos Int 2022; 33: 2233-2235. https://doi.org/10.1007/s00198-022-06486-9
- Brabnikova Maresova K. Secondary osteoporosis in patients with juvenile idiopathic arthritis. J Osteoporos 2011; 2011: 569417. https://doi.org/10.4061/2011/569417
- Simonini G, Giani T, Stagi S, de Martino M, Falcini F. Bone status over 1 yr of etanercept treatment in juvenile idiopathic arthritis. Rheumatology (Oxford) 2005; 44: 777-780. https://doi.org/10.1093/rheumatology/keh592
- Vojvodich PF, Hansen JB, Andersson U, Sävendahl L, Hagelberg S. Etanercept treatment improves longitudinal growth in prepubertal children with juvenile idiopathic arthritis. J Rheumatol 2007; 34: 2481-2485.
- Seriolo B, Paolino S, Sulli A, Cutolo M. Are there any positive effects of TNF-alpha blockers on bone metabolism? Reumatismo 2006; 58: 199-205. https://doi.org/10.4081/reumatismo.2006.199
- Kabala PA, Angiolilli C, Yeremenko N, et al. Endoplasmic reticulum stress cooperates with Toll-like receptor ligation in driving activation of rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Res Ther 2017; 19: 207. https://doi.org/10.1186/s13075-017-1386-x
- Matthews BG, Roeder E, Wang X, et al. Splenomegaly, myeloid lineage expansion and increased osteoclastogenesis in osteogenesis imperfecta murine. Bone 2017; 103: 1-11. https://doi.org/10.1016/j.bone.2017.06.004
- Spelling P, Bonfa E, Caparbo VF, Pereira RM. Osteoprotegerin/RANKL system imbalance in active polyarticular-onset juvenile idiopathic arthritis: a bone damage biomarker? Scand J Rheumatol 2008; 37: 439-444. https://doi.org/10.1080/03009740802116224
- Punzi L, Galozzi P, Luisetto R, et al. Post-traumatic arthritis: overview on pathogenic mechanisms and role of inflammation. RMD Open 2016; 2: e000279. https://doi.org/10.1136/rmdopen-2016-000279
- McDonagh JE. Osteoporosis in juvenile idiopathic arthritis. Curr Opin Rheumatol 2001; 13: 399-404. https://doi.org/10.1097/00002281-200109000-00010
- McKiernan FE. Musculoskeletal manifestations of mild osteogenesis imperfecta in the adult. Osteoporos Int 2005; 16: 1698-1702. https://doi.org/10.1007/s00198-005-1905-5
- Oz B, Olmez N, Memis A. Osteogenesis imperfecta: a case with hand deformities. Clin Rheumatol 2005; 24: 565-568. https://doi.org/10.1007/s10067-005-1087-8
Copyright and license
Copyright © 2025 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









