
Abstract
Background. Inflammatory myofibroblastic tumors (IMTs) are rare neoplasms in children. Traditionally, surgical resection has been the primary treatment modality with limited efficacy reported for conventional chemotherapy and radiation therapy. Recently, targeted therapies have emerged as potential options for selected cases. This study aimed to evaluate the demographic, clinical, laboratory, and radiological characteristics, as well as treatment outcomes, in children diagnosed with IMTs.
Methods. This study involved a retrospective review of medical records for eight children diagnosed with IMTs between 1990 and 2022. We collected demographic, clinical, laboratory, and radiological data, as well as treatment outcomes. Data on tumor characteristics, surgical procedures, and chemotherapy or targeted therapy treatments were extracted.
Results. The mean age at diagnosis was 9 years. None presented with metastatic disease at the time of diagnosis. Anaplastic lymphoma kinase (ALK) positivity was identified in tumor tissue from five patients. Among the six patients who underwent surgical resection, three achieved negative surgical margins. Of the three patients with positive surgical margins, one underwent re-resection, local and metastatic recurrences were noted in another, and one was started on crizotinib. A patient with an inoperable tumor at diagnosis was initiated on crizotinib and achieved complete remission. Ceritinib was administered to a patient with YWHAE-ROS fusion, resulting in more than 90% reduction in tumor volume. The median follow-up time was 67.5 months. The five-year overall survival and event-free survival rates for the cohort were 85.7% and 72.9%, respectively.
Conclusions. While surgical resection remains the cornerstone of treatment for IMTs, favorable outcomes can be achieved with chemotherapy and targeted therapies in selected cases. Increasing the utilization of targeted therapies may be beneficial, particularly through molecular studies aimed at minimizing the side effects associated with conventional chemotherapy.
Keywords: inflammatory myofibroblastic tumor, ALK inhibitor, crizotinib, ceritinib, childhood
Introduction
Inflammatory myofibroblastic tumors (IMTs) are rare tumors that typically occur in soft tissues, particularly in children and young adults, although they can arise at any age. They were previously known as inflammatory pseudotumors and are characterized by a proliferation of mesenchymal spindle cells along with a prominent inflammatory cell component. IMTs may lead to various clinical courses due to tumor size, localization and capacity to invade neighboring tissues. Epithelioid inflammatory myofibroblastic sarcoma (EIMS), which is a variant of IMT, is characterized by the proliferation of epithelioid and spindle-shaped myofibroblasts within a background of inflammatory cells. While IMTs are generally benign or low-grade tumors, EIMS presents with more aggressive features and a higher potential for recurrence and metastasis.1,2-4
Historically, surgical resection has been the primary treatment of IMTs, with little documented efficacy of traditional chemotherapy or radiation therapy.1-3,5
Comprehensive genomic analyses have identified various gene rearrangements, most notably involving the anaplastic lymphoma kinase (ALK) gene, which occurs in over 40% of IMT cases.6,7 Different patterns of ALK staining can be observed in both IMTs and EIMS, but the RANBP2-ALK fusion is specific to EIMS. Cases that do not exhibit ALK fusion are considerably less common and may involve translocations associated with ROS1, PDGFRB, NTRK3, RET, and IGF1R.8
In recent years, targeted therapies have been utilized for inoperable cases with identifiable targetable fusions. Therefore, the identification of specific molecular markers not only helps in diagnosing IMTs and differentiating them from other tumors with similar histological features but also is crucial for yielding new therapeutic targets and further refining existing treatment strategies. In this study, we share the treatment strategies applied to our patients diagnosed with IMT.
Materials and Methods
Medical records of children with IMT diagnosed and treated between 1990-2022 at the pediatric oncology clinic of a referral hospital were retrospectively reviewed. Demographic, clinical and radiological characteristics, treatment and outcome of the patients were evaluated. Follow-up time was recorded as the period from diagnosis to February 2023 or until the last visit. All patients were diagnosed histopathologically. In recent years, ALK has been investigated by immunocytochemistry (IHC) and ALK and other fusions were investigated by flourescence in situ hybridization (FISH) and real time polymerase chain reaction (RT-PCR).9
Non-mutilating surgical complete resection was performed whenever possible. In unresectable cases or in cases where surgery would lead to unacceptable morbidity, diagnosis was established by a tru-cut biopsy and neoadjuvant chemotherapy was initiated. In cases with incomplete resection, chemotherapy was used postoperatively. Chemotherapy consisted of adriamycin and ifosfamide.
The patients were evaluated by physical examination, complete blood count and biochemical tests before and during treatment and as necessary. None of the patients received radiation therapy.
This study was reviewed and approved by the Institutional Ethics Committee of İstanbul University, Oncology Institute (2023/1627454).
Statistical analyses
Statistics were calculated using IBM SPSS® 26 (Armonk, New York, U.S.). Kaplan-Meier method was used for survival analysis. Overall survival was calculated from the date of diagnosis to the date of last information on follow-up or death. Event-free survival was calculated from the date of diagnosis to the date of the first event, such as progression, relapse, or death from any cause.
Results
Patient characteristics are summarized in Table I. There were eight patients (5 male, 3 female), with a mean age at diagnosis of 9 years (range: 8 months to 17 years). Six patients had IMT and two had EIMS. None of the patients presented with metastatic disease at the time of diagnosis.
|
*The patient with a YWHAE-ROS fusion. ALK: anaplastic lymphoma kinase AWD: alive with disease, EMS: Epithelioid inflammatory myofibroblastic sarcoma, F: female, IA: ifosfamide, adriamycine, ICE: ifosfamide, carboplatinum, etoposide, IMT: Inflammatory myofibroblastic tumor, M: male, NED: no evidence of disease, VAC: vincristine, adriamycine, cyclophosphamide. |
|||||||||||
| Table I. Patients’ characteristics | |||||||||||
| Age/Sex | Histopathology | Localization | ALK status | Metastasis at diagnosis | Treatment | Surgical margin | Recurrence / Progression | Further treatment | Latest status | Follow-up period (months) | Event-free survival (months) |
| 12 y/M | IMT | Intestine | Positive | No | Resection | Negative | No | NED | 163 | 163 | |
| 10 y/M | IMT | Retroperitoneum | Positive | No | Resection | Positive | Local and metastatic (lung, omentum, liver, bone) |
4 courses with VAC, and 12 courses with ICE after progression with VAC | EX | 14 | - |
| 8 y/M | EMS | Back | Unknown | No | Resection | First positive. Negative after re-resection | No | NED | 124 | 124 | |
| 7 y/M | IMT | Retroperitoneum | Positive | No | Crizotinib after resection for 12 mo. | Positive | No | NED | 99 | 98 | |
| 12 y/F | IMT | Right lower extremity | Positive | No |
Crizotinib for 7 mo. |
Only biopsy | Local (2 mo after stopping crizotinib) |
Crizotinib | NED | 23 | 4 |
| 7 y/M | IMT | Liver | Unknown | No | Resection | Negative | No | NED | 174 | 174 | |
| 8 mo/F | EMS | Intestine | Positive | No | Resection | Negative | No | NED | 36 | 35 | |
| 17 y/F* | IMT | Right lower extremity | Negative* | No | Resection | Negative | Local (At 16 mo) |
One course with IA and ceritinib after toxicity with chemotherapy | AWD | 9 | 4 |
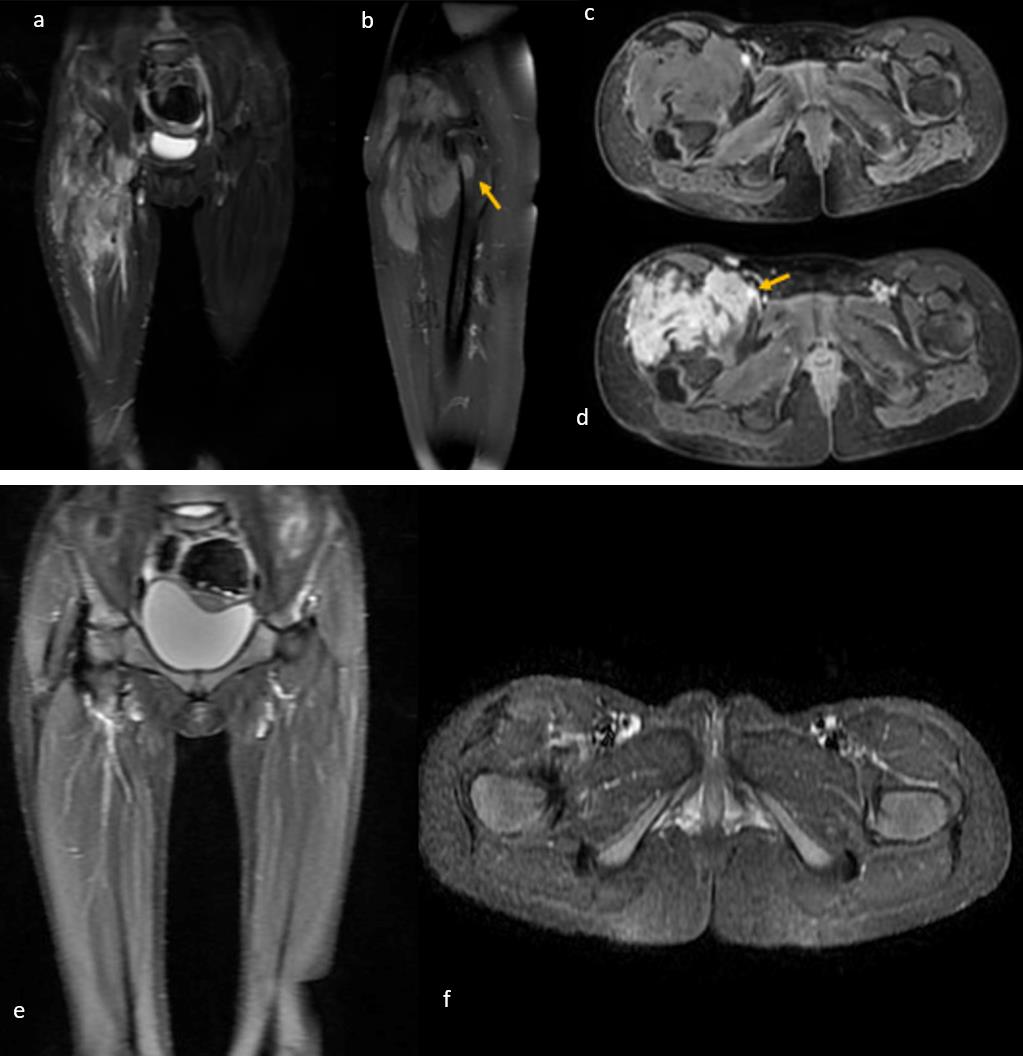
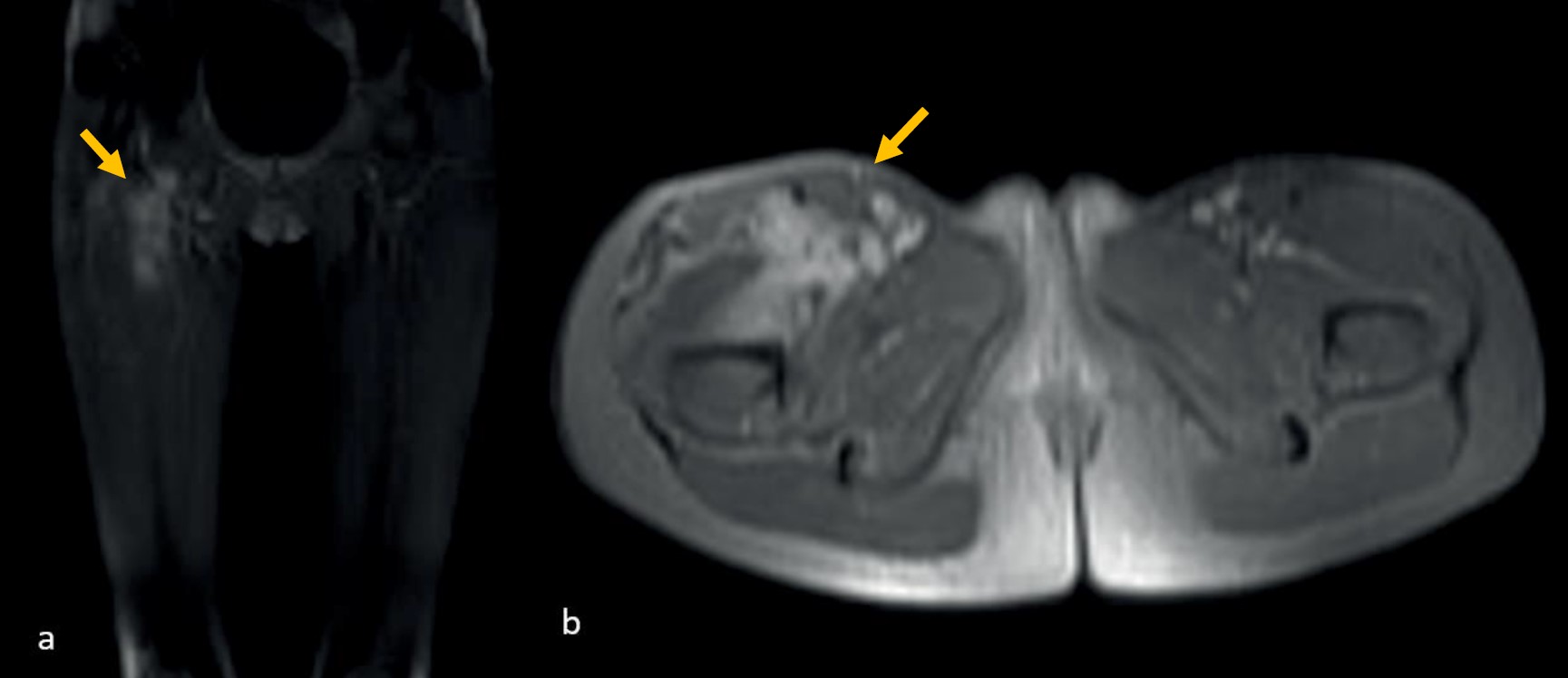
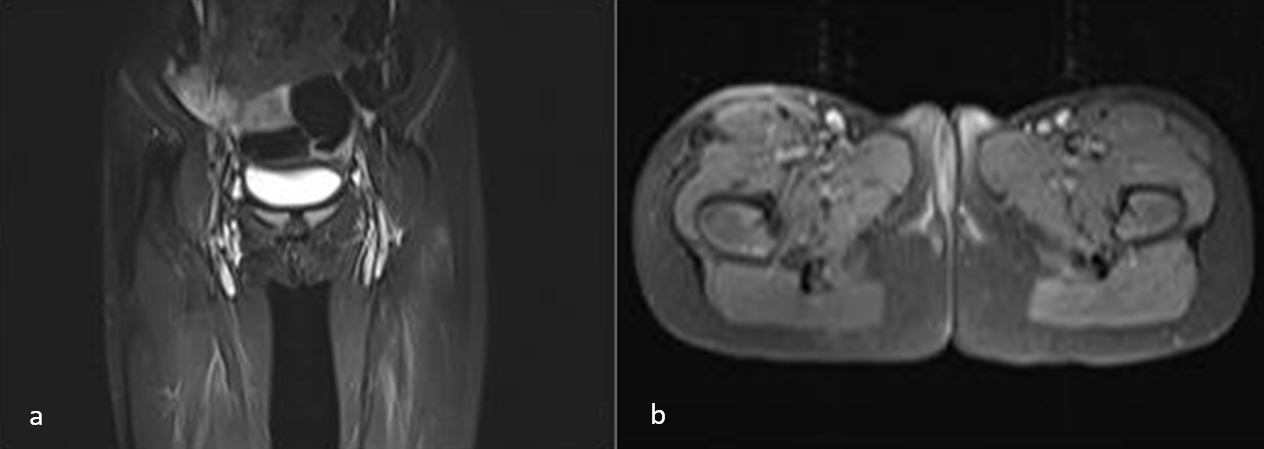
Three of the six patients (including one with EIMS) who underwent surgical resection alone achieved negative surgical margins and are currently under follow-up with no evidence of disease (NED). Among the other two patients who underwent surgical resection alone, both had positive surgical margins. One of these patients underwent re-resection for EIMS and is currently under follow-up without any events. A patient who underwent surgical resection at diagnosis and had positive surgical margins received crizotinib treatment for one year and is currently under follow-up in complete remission. Crizotinib treatment was initiated for a patient diagnosed with an inoperable tumor. After seven months of treatment, the patient achieved complete remission as confirmed by magnetic resonance imaging (MRI) (Fig. 1). Following evaluation by the multidisciplinary tumor board, it was decided to discontinue the treatment. However, local recurrence was detected on MRI two months after the cessation of crizotinib (Fig. 2). Although the patient reported no complaints and exhibited no significant findings on physical examination, crizotinib was restarted due to the infiltrative nature of the tumor and the potential morbidities associated with surgical intervention. Two months after resuming crizotinib, MRI showed a complete response (Fig. 3). The patient has been continuing treatment for four months.
Another patient diagnosed with ALK-negative IMT, who had undergone surgical resection with negative margins, was subsequently treated with conventional chemotherapy consisting of ifosfamide and doxorubicin following disease recurrence. The patient presented with symptoms including cachexia, fever, and hypercalcemia after the recurrence of the disease. The etiological factors for the fever and hypercalcemia were investigated, and it was ultimately determined that these symptoms might be related to malignancy. Despite treatment with intravenous fluids and furosemide, the hypercalcemia remained refractory. After the first cycle of chemotherapy, the patient developed febrile neutropenia, which progressed to life-threatening acute respiratory distress syndrome (ARDS), necessitating 10 days of intensive care and the hypercalcemia was refractory to chemotherapy. Molecular analysis of the tumor tissue via next-generation sequencing (NGS) revealed the presence of a YWHAE-ROS fusion. Consequently, the patient was initiated on ceritinib treatment, which has demonstrated comparable activity to crizotinib against ROS1. As a result, treatment with ceritinib was initiated, which has shown comparable efficacy to crizotinib against ROS1. Within the first week of treatment, hypercalcemia improved, and the patient’s appetite significantly increased by the second week. Remarkably, the previously bedridden patient was able to mobilize. A follow-up MRI conducted three months post-initiation of treatment demonstrated a reduction of over 90% in tumor size. The patient has continued on ceritinib for six months. The patient has been using ceritinib for 6 months.
The median follow-up time for the entire cohort was 67.5 months (range: 9–174 months). The five-year overall survival and event-free survival rates for the group were 85.7% and 72.9%, respectively.
Discussion
Inflammatory myofibroblastic tumors, first described by Brunn et al. in 1939, are rare neoplasms that primarily occur in the lungs, abdomen, and orbit.10,11 They most commonly present within the first two decades of life, although they can arise at any age and can vary widely in size and location.1-4 Histologically benign, IMTs may exhibit locally aggressive behavior and, in rare cases, can metastasize.3,5
The primary treatment for IMTs has historically been complete surgical resection, although this can often be challenging and associated with significant morbidity. Incomplete resection has been linked to a high recurrence rate. Other treatment options, such as nonsteroidal anti-inflammatory drugs (NSAIDs), high-dose corticosteroids, chemotherapy, and radiotherapy, may carry serious side effects and their efficacy remains unclear.2,3
Approximately half of the observed IMTs involve a clonal translocation that activates the ALK receptor tyrosine kinase gene located at chromosome band 2p23, in conjunction with various partner genes. The frequency of ALK rearrangements is notably high among pediatric and young adult patients with IMT.6,12
In cases of IMTs that are negative for ALK fusion genes, other fusion genes such as c-ros oncogene 1 (ROS1), neurotrophin tropomyosin receptor kinase (NTRK), platelet-derived growth factor receptor (PDGFR), and RET have been identified.9 Additionally, certain partner genes associated with ALK or other fusion genes may correlate with clinical features. For example, EIMS is a subtype of IMT that exhibits aggressive behavior and is often associated with specific fusion genes like RANBP2-ALK and RRBP1-ALK. While numerous potential fusion genes have been identified in IMT, conducting gene panel testing at the initial stages can be cost-prohibitive. If the IHC method can help narrow down the candidates for fusion genes, it could be advantageous in reducing both the cost and time required for diagnosis.13-16
Following the study by Mosse et al.17 indicating the effectiveness and safety of crizotinib in pediatric patients with ALK-positive tumors, we began to consider its use in patients with inoperable tumors where ALK status was known at the time of diagnosis, as well as in cases with positive surgical margins. In one of the patients in this study, complete remission was achieved after seven months of crizotinib used as neoadjuvant therapy, leading to the discontinuation of treatment. However, a local recurrence was noted two months later, and a complete response was achieved two months after resuming crizotinib.
According to the existing literature, responses to crizotinib can vary among tumors, and there is no definitive guidance on the optimal duration of treatment for patients who respond positively.18-27 Tumor regression is often observed early in the treatment course.4,18 In a study involving eight cases of ALK-positive IMTs, crizotinib was discontinued in five patients after a median treatment duration of one year (range: 0.2 to 3.0 years). These patients were subsequently followed for a median of 1.7 years (range: 0.3 to 3.7 years), during which four achieved complete remission (CR) and one had stable disease (SD). This suggests that treatment may be safely discontinued without a rapid recurrence, in contrast to observations in anaplastic large cell lymphoma.17,28
Disease progression or recurrence has also been reported both during and after the cessation of crizotinib treatment. Cases of progression or recurrence occurring during or after crizotinib treatment are summarized in Supplementary Table 1.25-34
Following the previous reports by Lovly et al.12 and Comandini et al.35, here we represent the third case in the literature carrying YWHAE1-ROS1 fusion detected by NGS. Ceritinib, a selective oral tyrosine kinase inhibitor (TKI) of ALK, operates similarly to crizotinib but lacks MET-inhibiting capabilities and received FDA approval for treating ALK-positive non-small cell lung cancer (NSCLC) resistant to crizotinib. In enzymatic assays, ceritinib demonstrated 20 times greater potency against ALK compared to crizotinib and exhibited comparable efficacy against ROS. Additionally, ceritinib exhibited comparable efficacy to crizotinib against ROS1. In an open-label multicenter phase II study, ceritinib demonstrated robust clinical activity in NSCLC patients with ROS1 rearrangement, achieving a 62% objective response rate. The median progression-free survival was 9.3 months for all patients and 19.3 months for those who were crizotinib-naïve.36-38 Ceritinib has also exhibited clinical effectiveness in patients with IMT. Li et al.39 reported the first instance of using ceritinib to treat a ROS1-rearranged IMT, resulting in a partial response. By opting for ceritinib in our case, we were able to avoid the severe side effects associated with chemotherapy while achieving a high success rate in a remarkably short period.
This study has a few limitations. The small sample size (eight patients) limits generalizability, and the retrospective design introduces potential biases in treatment protocols and follow-up care. Not all patients underwent comprehensive molecular profiling, which could have missed other therapeutic targets. Additionally, the lack of long-term data on the side effects of targeted therapies like crizotinib and ceritinib is a significant gap. Lastly, a direct comparison between traditional chemotherapy and targeted therapies was not included.
Future studies should aim to include larger, multi-center cohorts to validate these findings. Expanding molecular profiling using next-generation sequencing could uncover additional therapeutic targets. Long-term studies are needed to assess the durability and safety of targeted therapies, and research should focus on identifying optimal treatment durations. Prospective trials comparing targeted therapies with conventional chemotherapy regimens would provide valuable insights into the best treatment approaches for IMTs.
In conclusion, the identification of molecular alterations in rare malignancies, such as IMTs, is essential for guiding personalized treatment strategies with targeted therapies. Tailoring treatment based on specific molecular profiles allows for the use of TKIs, which have demonstrated significant efficacy while often reducing the adverse effects associated with conventional chemotherapy. This personalized approach not only enhances treatment outcomes but also improves the overall quality of life for patients. By prioritizing a comprehensive diagnostic workup and focusing on individualized treatment plans, we can ensure that each patient receives the most appropriate and effective care, ultimately advancing the management of rare oncological conditions.
The authors declare that there is no conflict of interest to disclose.
Acknowledgements
We thank Assoc. Prof. Dr. Ahmet Salduz, İstanbul University, İstanbul Faculty of Medicine, Department of Orthopedics and Traumatology; Prof. Dr. Gökçen Ünverengil, İstanbul University, İstanbul Faculty of Medicine, Department of Pathology; Prof. Dr. Sebuh Kuruoglu, İstanbul University-Cerrahpaşa, Department of Radiology for orthopedics, pathology and radiology consultations.
Ethical approval
This study was approved by İstanbul University, Oncology Institute Ethics Committee (13.02.2023-1627454). Informed consent was obtained from the parents.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Karnak I, Senocak ME, Ciftci AO, et al. Inflammatory myofibroblastic tumor in children: diagnosis and treatment. J Pediatr Surg 2001; 36: 908-912. https://doi.org/10.1053/jpsu.2001.23970
- Theilen TM, Soerensen J, Bochennek K, et al. Crizotinib in ALK+ inflammatory myofibroblastic tumors-current experience and future perspectives. Pediatr Blood Cancer 2018; 65: e26920. https://doi.org/10.1002/pbc.26920
- Ding Y, Yang HY, Zhang D, et al. Diagnosis and treatment of inflammatory myofibroblastoma in children and adolescents. Chin Med J (Engl) 2019; 132: 1110-1112. https://doi.org/10.1097/CM9.0000000000000176
- Mossé YP, Voss SD, Lim MS, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a Children’s Oncology Group Study. J Clin Oncol 2017; 35: 3215-3221. https://doi.org/10.1200/JCO.2017.73.4830
- Janik JS, Janik JP, Lovell MA, Hendrickson RJ, Bensard DD, Greffe BS. Recurrent inflammatory pseudotumors in children. J Pediatr Surg 2003; 38: 1491-1495. https://doi.org/10.1016/s0022-3468(03)00501-3
- Antonescu CR, Suurmeijer AJ, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 gene fusions and rare novel RET rearrangement. Am J Surg Pathol 2015; 39: 957-967. https://doi.org/10.1097/PAS.0000000000000404
- Griffin CA, Hawkins AL, Dvorak C, Henkle C, Ellingham T, Perlman EJ. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res 1999; 59: 2776-2780.
- Sommer S, Schmutz M, Schaller T, et al. Individualized targeted treatment in a case of a rare TFG::ROS1 fusion positive inflammatory myofibroblastic tumor (IMT). Cancer Rep (Hoboken) 2024; 7: e1916. https://doi.org/10.1002/cnr2.1916
- Yamamoto H, Yoshida A, Taguchi K, et al. ALK, ROS1 and NTRK3 gene rearrangements in inflammatory myofibroblastic tumours. Histopathology 2016; 69: 72-83. https://doi.org/10.1111/his.12910
- Brunn H. Two interesting benign lung tumors of contradictory histopathology: remarks on the necessity for maintaining the chest tumor registry. Journal of Thoracic Surgery 9: 119-131, 1939. https://doi.org/10.1016/S0096-5588(20)32030-4
- Cakir E, Cakir FB, Bingol D, Gedik AH, Soysal O. Not all that wheezes is asthma or foreign body aspiration: endobrochial inflammatory myofibroblastic tumor. Indian J Pediatr 2014; 81: 306-307. https://doi.org/10.1007/s12098-013-1318-y
- Lovly CM, Gupta A, Lipson D, et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov 2014; 4: 889-895. https://doi.org/10.1158/2159-8290.CD-14-0377
- Takeuchi K, Soda M, Togashi Y, et al. Pulmonary inflammatory myofibroblastic tumor expressing a novel fusion, PPFIBP1-ALK: reappraisal of anti-ALK immunohistochemistry as a tool for novel ALK fusion identification. Clin Cancer Res 2011; 17: 3341-3348. https://doi.org/10.1158/1078-0432.CCR-11-0063
- Pickett JL, Chou A, Andrici JA, et al. Inflammatory myofibroblastic tumors of the female genital tract are under-recognized: a low threshold for ALK immunohistochemistry is required. Am J Surg Pathol 2017; 41: 1433-1442. https://doi.org/10.1097/PAS.0000000000000909
- Yamamoto H, Nozaki Y, Kohashi K, Kinoshita I, Oda Y. Diagnostic utility of pan-Trk immunohistochemistry for inflammatory myofibroblastic tumours. Histopathology 2020; 76: 774-778. https://doi.org/10.1111/his.14010
- Mariño-Enríquez A, Wang WL, Roy A, et al. Epithelioid inflammatory myofibroblastic sarcoma: An aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am J Surg Pathol 2011; 35: 135-144. https://doi.org/10.1097/PAS.0b013e318200cfd5
- Mosse YP, Lim MS, Voss SD, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol 2013; 14: 472-480. https://doi.org/10.1016/S1470-2045(13)70095-0
- Craig E, Wiltsie LM, Beaupin LK, et al. Anaplastic lymphoma kinase inhibitor therapy in the treatment of inflammatory myofibroblastic tumors in pediatric patients: case reports and literature review. J Pediatr Surg 2021; 56: 2364-2371. https://doi.org/10.1016/j.jpedsurg.2021.02.004
- Jindal A, Bal A, Agarwal R. Inflammatory myofibroblastic tumor of the trachea in the pediatric age group: case report and systematic review of the literature. J Bronchology Interv Pulmonol 2015; 22: 58-65. https://doi.org/10.1097/LBR.0000000000000105
- Rafee S, Elamin YY, Joyce E, et al. Neoadjuvant crizotinib in advanced inflammatory myofibroblastic tumour with ALK gene rearrangement. Tumori 2015; 101: e35-e39. https://doi.org/10.5301/tj.5000245
- Suleymanova A, Imyanitov E, Roschin V, et al. Rapid response to crizotinib in 3-year-old boy with ROS1-rearranged inflammatory myofibroblastic tumor of the stomach. In: Pediatric Blood & Cancer. USA: Wiley; 2018: S389-S389.
- Ray S, Willis C, Murphy D. Crizotinib therapy in anaplastic lymphoma kinase-positive inflammatory myofibroblastic tumour: when to stop? In: Pediatric Blood & Cancer. USA: Wiley; 2018: S398-S398.
- Arakawa A, Yonemori K, Kumamoto T, et al. Successful treatment of a highly aggressive abdominal cavity ALK-rearranged inflammatory myofibroblastic tumor with alectinib: a case report. In: Pediatric Blood & Cancer. USA: Wiley; 2017: S40.
- Sugawa M, Watanabe K, Arakawa Y, et al. Inflammatory myofibroblastic tumor of the liver with a remarkable response to crizotinib in a young child. In: Pediatric Blood & Cancer. USA: Wiley; 2017: S40-S41.
- Gaudichon J, Jeanne-Pasquier C, Deparis M, et al. Complete and repeated response of a metastatic ALK-rearranged inflammatory myofibroblastic tumor to crizotinib in a teenage girl. J Pediatr Hematol Oncol 2016; 38: 308-311. https://doi.org/10.1097/MPH.0000000000000498
- Kiratli H, Uzun S, Varan A, Akyüz C, Orhan D. Management of anaplastic lymphoma kinase positive orbito-conjunctival inflammatory myofibroblastic tumor with crizotinib. J AAPOS 2016; 20: 260-263. https://doi.org/10.1016/j.jaapos.2016.01.009
- Sarmiento DE, Clevenger JA, Masters GA, et al. Epithelioid inflammatory myofibroblastic sarcoma: a case report. J Thorac Dis 2015; 7: E513-E516.
- Trahair T, Gifford AJ, Fordham A, et al. Crizotinib and surgery for long-term disease control in children and adolescents with ALK-positive inflammatory myofibroblastic tumors. JCO Precis Oncol 2019; 3: PO.18.00297. https://doi.org/10.1200/PO.18.00297
- Mansfield AS, Murphy SJ, Harris FR, et al. Chromoplectic TPM3-ALK rearrangement in a patient with inflammatory myofibroblastic tumor who responded to ceritinib after progression on crizotinib. Ann Oncol 2016; 27: 2111-2117. https://doi.org/10.1093/annonc/mdw405
- Michels SYF, Scheel AH, Wündisch T, et al. ALK(G1269A) mutation as a potential mechanism of acquired resistance to crizotinib in an ALK-rearranged inflammatory myofibroblastic tumor. NPJ Precis Oncol 2017; 1: 4. https://doi.org/10.1038/s41698-017-0004-3
- Yuan C, Ma MJ, Parker JV, Mekhail TM. Metastatic anaplastic lymphoma kinase-1 (ALK-1)-rearranged inflammatory myofibroblastic sarcoma to the brain with leptomeningeal involvement: favorable response to serial ALK inhibitors: a case report. Am J Case Rep 2017; 18: 799-804. https://doi.org/10.12659/ajcr.903698
- Alan O, Kuzhan O, Koca S, et al. How long should we continue crizotinib in ALK translocation-positive inflammatory myofibroblastic tumors? Long-term complete response with crizotinib and review of the literature. J Oncol Pharm Pract 2020; 26: 1011-1018. https://doi.org/10.1177/1078155219879757
- Schöffski P, Kubickova M, Wozniak A, et al. Long-term efficacy update of crizotinib in patients with advanced, inoperable inflammatory myofibroblastic tumour from EORTC trial 90101 CREATE. Eur J Cancer 2021; 156: 12-23. https://doi.org/10.1016/j.ejca.2021.07.016
- Butrynski JE, D’Adamo DR, Hornick JL, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med 2010; 363: 1727-1733. https://doi.org/10.1056/NEJMoa1007056
- Comandini D, Catalano F, Grassi M, et al. Outstanding response in a patient with ROS1-Rearranged inflammatory myofibroblastic tumor of soft tissues treated with crizotinib: case report. Front Oncol 2021; 11: 658327. https://doi.org/10.3389/fonc.2021.658327
- Shaw AT, Engelman JA. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med 2014; 370: 2537-2539. https://doi.org/10.1056/NEJMc1404894
- Facchinetti F, Loriot Y, Kuo MS, et al. Crizotinib-resistant ROS1 mutations reveal a predictive kinase inhibitor sensitivity model for ROS1- and ALK-rearranged lung cancers. Clin Cancer Res 2016; 22: 5983-5991. https://doi.org/10.1158/1078-0432.CCR-16-0917
- Lim SM, Kim HR, Lee JS, et al. Open-label, multicenter, phase ii study of ceritinib in patients with non-small-cell lung cancer harboring ROS1 rearrangement. J Clin Oncol 2017; 35: 2613-2618. https://doi.org/10.1200/JCO.2016.71.3701
- Li Y, Chen X, Qu Y, et al. Partial response to ceritinib in a patient with abdominal inflammatory myofibroblastic tumor carrying a TFG-ROS1 fusion. J Natl Compr Canc Netw 2019; 17: 1459-1462. https://doi.org/10.6004/jnccn.2019.7360
Copyright and license
Copyright © 2025 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









