
Abstract
Background. Citrin deficiency (CD), caused by mutations in the SLC25A13 gene, is a rare autosomal recessive urea cycle disorder with variable clinical presentations depending on age. These include neonatal intrahepatic cholestasis (NICCD), failure to thrive with dyslipidemia, and adult-onset type II citrullinemia. Patients with NICCD typically present with transient intrahepatic cholestasis in infancy, which often resolves spontaneously by one year of age; however, some may progress to severe complications later in life.
Case Presentation. Four cases diagnosed with NICCD phenotype are presented. All patients presented with neonatal cholestasis, hypertransaminasemia, galactosuria, and elevated citrulline levels. Molecular analysis identified three disease-causing variants: two previously reported variants, c.955C>T (p.Arg319*) and c.74C>A (p.Ala25Glu), and a novel variant, c.1359G>T (p.Lys453Asn). Treatment included a galactose-free formula, medium-chain triglycerides, and nutritional supplementation, resulting in biochemical and clinical improvement. All patients in our series exhibited a milder clinical course, with no episodes of hyperammonemia or hypoglycemia, no progression to liver failure, and favorable long-term outcomes with dietary management. During a long-term follow-up period ranging from 7 to 11 years, no severe complications were observed. Notably, one patient developed a recurrence of cataract, emphasizing the importance of lifelong dietary adherence and regular eye examinations.
Conclusions. The findings in this paper further expand the genotypic spectrum and genotype-phenotype correlations of CD. Lifelong follow-up is recommended, including ocular examination.
Keywords: citrin deficiency, cholestasis, SLC25A13, citrullinemia type 2, urea cycle disorder
Introduction
Citrin, a calcium-binding aspartate/glutamate carrier, is a component of the malate–aspartate shuttle and is closely linked to several biochemical pathways, including glycolysis, gluconeogenesis, de novo lipogenesis, beta-oxidation, the tricarboxylic acid (TCA) cycle, and the urea cycle. Citrin deficiency (CD), an autosomal recessive trait, is a rare disorder of the urea cycle caused by mutations in the SLC25A13 gene. It was first described in the Japanese population and is quite common among East Asians, but it is now considered a pan-ethnic disorder.1 The clinical spectrum of CD includes three distinct age-related phenotypes: neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD, OMIM#605814), failure to thrive (FTT) and dyslipidemia by citrin deficiency (FTTDCD), and adult-onset type II citrullinemia (CTLN2, OMIM #603471).2,3 NICCD patients primarily present with transient intrahepatic cholestasis during the infantile period; additional manifestations include hepatomegaly, citrullinemia, ketotic hypoglycemia, elevated levels of certain amino acids, hypoalbuminemia, cataracts and developmental delay.1 In most NICCD patients, symptoms resolve spontaneously by 12 months of age. However, in rare cases, severe hepatic failure may occur, necessitating liver transplantation. Following a silent remission period until after adolescence, less than 20% of patients develop a fatal metabolic disease, CTLN2, which is distinguished by recurrent episodes of hyperammonemia, hepatosteatosis, and neuropsychiatric manifestations such as disorientation, delirium, cognitive impairment, and abrupt episodes of unconsciousness.2,4 The onset of symptoms can be sudden and is typically observed between the ages of 20 and 40 years.4
In this study, we present clinical and laboratory findings and outcomes of four genetically confirmed cases of NICCD patients from non-Asian origin. Our report expands the mutation spectrum of the SLC25A13 gene with the identification of a novel c.1359G>T (p.Lys453Asn) variant and highlights clinical observations such as cataract recurrence during follow-up. This study aimed to highlight the importance of disease awareness, clinical follow-up during early infancy and childhood, and the importance of ocular examinations in the follow-up of cases with CD.
Case Presentations
We present the clinical characteristics of a case series diagnosed with NICCD, including three siblings from the same family, comprising a set of dizygotic twins, and an additional patient from a different family. All the studies were conducted in accordance with the Declaration of Helsinki and guidelines for good clinical practice. All legal guardians of the patients were informed, and their informed consent was obtained.
Case A1
A 17-day-old male infant, whose parents were second cousins, was referred to our metabolic center for abnormal newborn screening test for phenylketonuria (PKU). In medical history, the patient, born at 38 weeks of gestation, had a birth weight of 2290 g (-2.6 SD), a length of 44 cm (-2.6 SD), and a head circumference of 31 cm (-2.8 SD).
The newborn screening performed on the second day was normal (0.1 mg/dL; cut-off: 2) but repeated phenylalanine (Phe) level on the tenth day of life was 5.5 mg/dL. At our center laboratory tests had revealed normal Phe and elevated citrulline (Cit) levels (372 µmol/L, normal range: 3-57) by tandem mass spectrometry (MS). Evaluation for elevated Cit included further investigations for distal urea cycle disorders and CD. At the initial admission, icterus with pale colored stools and mild hepatomegaly were detected. No dysmorphic appearance was noted. He subsequently developed progressive cholestasis and worsening jaundice by 47 days of age. Laboratory results revealed cholestasis, elevated international normalized ratio (INR), hypertransaminasemia, significantly elevated alpha-fetoprotein (AFP), positive urine test for reducing substances, and galactosuria (Table I). Stool examination detected +3 steatorrhea. Fundus examination was normal, but cataract formation was identified. Echocardiography was normal, and neuromotor development progressed age-appropriately. Quantitative plasma amino acid analysis was consistent with CD, as characteristic alterations in blood amino acid levels were observed. These included increased levels of Cit (376 μmol/L; reference range [RR]: 6–35), arginine (Arg, 191.2 μmol/L; RR: 18–102), methionine (Met, 124.8 μmol/L; RR: 9–44), and threonine (Thr, 685 μmol/L; RR: 33–160), as well as an elevated threonine-to-serine ratio (Thr/Ser, 4; RR: <1.1) (Table II). Genetic analyses revealed homozygosity for the pathogenic c.955C>T (p.Arg319*) variant at exon 10 of the SLC25A13 gene. Both parents were confirmed to be heterozygous for the variant. The patient was prescribed a galactose-free formula with medium-chain triglycerides (MCT), and Arg and fat-soluble vitamin supplements were initiated. While the clinical features improved, the elevation in Cit persisted until the age of 25 months. After achieving clinical and biochemical improvement, the galactose-free and MCT diet was discontinued at two years of age, recommending avoidance of excessive galactose content in the diet. Although previously normal, an abdominal ultrasound displayed grade 1 hepatosteatosis at the age of six years. On the latest visit to the outpatient clinic, at the age of eleven, the patient weighed 33 kg (-0.6 SD) and measured 141 cm (-0.4 SD). Physical examination findings were normal, and all biochemical parameters, including liver enzymes and synthetic functions, Cit level, lipid profiles, AFP, and ammonia, were within the normal range. Grade 1 hepatic steatosis persisted according to ultrasonography (USG). Ophthalmologic examination was completely normal, with no evidence of cataracts or other ocular abnormalities.
| * The values represent the measurements at the time of diagnosis. ALT, alanine aminotransferase; AST, aspartate aminotransferase; AFP, alpha-feto protein; d, days; F, female; g, gram; INR, international normalized ratio; N, normal; NA, not available; NICCD, neonatal intrahepatic cholestasis by citrin deficiency; M, male; mo, month; SF, symptom free; w, week; y, year. | |||||||||||
| Table I. Summary of our patients (Patients 1-4) and other reported patients with NICCD from Türkiye. | |||||||||||
| Characteristic |
|
|
|
|
|
|
|
|
|
|
|
| Age |
|
|
|
|
|
|
|
|
|
|
|
| Gender |
|
|
|
|
|
|
|
|
|
|
|
| Gestation week, birth weight |
|
|
|
|
|
|
|
|
|
|
|
| Age at symptom onset |
|
|
|
|
|
|
|
|
|
|
|
| Clinical presentation |
|
|
|
|
|
|
|
|
|
|
|
| Biochemical findings* | |||||||||||
| Total bilirubin, mg/dl |
|
|
|
|
|
|
|
|
|
|
|
| Direct bilirubin, mg/dl |
|
|
|
|
|
|
|
|
|
|
|
| AST, U/L |
|
|
|
|
|
|
|
|
|
|
|
| ALT, U/L |
|
|
|
|
|
|
|
|
|
|
|
| Hyperammonemia |
|
|
|
|
|
|
|
|
|
|
|
| INR |
|
|
|
|
|
|
|
|
|
|
|
| Hypoglycemia |
|
|
|
|
|
|
|
|
|
|
|
| Albumin, g/dl |
|
|
|
|
|
|
|
|
|
|
|
| AFP, ng/ml |
|
|
|
|
|
|
|
|
|
|
|
| Galactosuria |
|
|
|
|
|
|
|
|
|
|
|
| SLC25A13 genotype |
|
|
|
|
|
|
|
|
|
|
|
| Outcome |
|
|
|
|
|
|
|
|
|
|
|
| Reference |
|
|
|
|
|
|
|
|
|
|
|
| Abnormal values are in bold. | ||||
| Table II. Plasma quantitative amino acid levels of the patients at diagnosis (μmol/L). | ||||
| Plasma level (Reference range) |
|
|
|
|
| Citrulline (6-35) |
|
|
|
|
| Threonine (33-160) |
|
|
|
|
| Methionine (9-44) |
|
|
|
|
| Arginine (18-102) |
|
|
|
|
| Tyrosine (14-114) |
|
|
|
|
| Lysine (56-200) |
|
|
|
|
| Glutamine (368-652) |
|
|
|
|
| Isoleucine (28-92) |
|
|
|
|
| Leucine (55-149) |
|
|
|
|
| Phenylalanine (26-120) |
|
|
|
|
| Tryptophan (23-70) |
|
|
|
|
| Valine (79-267) |
|
|
|
|
| Histidine (30-110) |
|
|
|
|
| Threonine/Serine (< 1.1) |
|
|
|
|
Case A2
Case A2, the dizygotic twin brother of Case A1, was delivered at 38 weeks of gestation, weighing 2160 g (-3.0 SD), with a length of 45 cm (-2.1 SD) and a head circumference of 31 cm (-2.8 SD). At the age of 17 days, the infant was referred for abnormal newborn screening result for PKU. Subsequent tandem MS testing revealed normal Phe and elevated Cit levels (352 µmol/L, RR: 3-57). At the age of 47 days, the patient presented with jaundice. Physical examination revealed minimal hepatomegaly. Laboratory work-up revealed cholestasis, elevated INR, and hypertransaminasemia, elevated AFP, positive urine test for reducing substances (Table I). Screening for classical galactosemia with the Beutler test was unremarkable. Quantitative plasma amino acids were consistent with CD (Table II). Ophthalmologic examination revealed a posterior subcapsular cataract. Abdominal USG was normal. The patient was prescribed a galactose-free formula with MCT, and supplemented with Arg and fat-soluble vitamin supplements as his twin brother. Sequence analysis of the SCL25A13 gene revealed the same novel homozygous variant c.955C>T (p.Arg319*).
After 25 months, all biochemical tests were normalized, the cataract was resolved, and the galactose-free MCT-containing diet was discontinued, which was replaced by a low-galactose diet. Throughout the patient’s follow-up visits, reducing substances in urine were consistently negative. Nevertheless, at the age of six, a mild cataract development was detected during the eye examination. Abdominal USG was normal. The patient was advised to resume the galactose-free dietary treatment. His current diet includes a protein: fat: carbohydrate ratio of 16%: 48%: 36%, along with the appropriate energy intake, in addition to the galactose-free diet with MCT oil. At the age of eight and a half years, the Wechsler Intelligence Scale for Children – Revised (WISC-R) disclosed an intelligence quotient (IQ) of 104 (verbal IQ, 95; performance IQ, 114). He was 11 years old at the last outpatient evaluation and his weight was 33 kg (-0.7 SD), and his height measured 143 cm (-0.1 SD). Physical examination findings were normal, and all biochemical parameters, including liver enzymes (aspartate and alanine aminotransferases) and synthetic functions (albumin level and coagulation parameters such as INR), Cit level, lipid profiles, AFP, and ammonia, were within the normal range. In the abdominal USG, no hepatic steatosis was detected. The cataract had not disappeared or progressed, but remained stable with dietary intervention of galactose restriction.
Case A3
This patient, the younger sister of patients A1 and A2, was delivered at 33+5 weeks of gestation due to preterm labor, with a birth weight of 1620 g (-0.5 SD), a length of 45 cm (0.8 SD), and a head circumference of 25 cm (-3.3 SD). Due to prematurity and respiratory distress, the patient was admitted to the neonatal intensive care unit. At 25 days of age, the patient remained clinically stable in terms of respiratory status; however, cholestasis was noted, and the urine test was positive for reducing substances. On physical examination, mild jaundice of the skin and bulbar conjunctiva were detected whereas no hepatosplenomegaly was noticed (Table I). The infant remained hospitalized for 37 days, with the highest recorded ammonia level reaching 96 µg/dL (normal range <110 µg/dL). The patient was prescribed a galactose-free formula and cholestasis resolved. The SLC25A13 variant was confirmed and identified as the same novel homozygous variant, c.955C>T (p.Arg319*), as in her brothers. After achieving clinical and biochemical improvement at the age of three years, the patient’s diet was eased which excluded milk and dairy products. Denver Developmental Screening Test II at age five years was age-appropriate. The patient is following a diet with a protein: fat: carbohydrate ratio of 16%: 43%: 41%, with an energy intake appropriate for her age. On the last admission to the outpatient clinic, at the age of seven years, the patient weighed 23 kg (-0.1 SD) and measured 116 cm (-1.1 SD) in length. Physical examination findings, abdominal USG, eye examination, and blood tests including liver function tests were normal.
Case B1
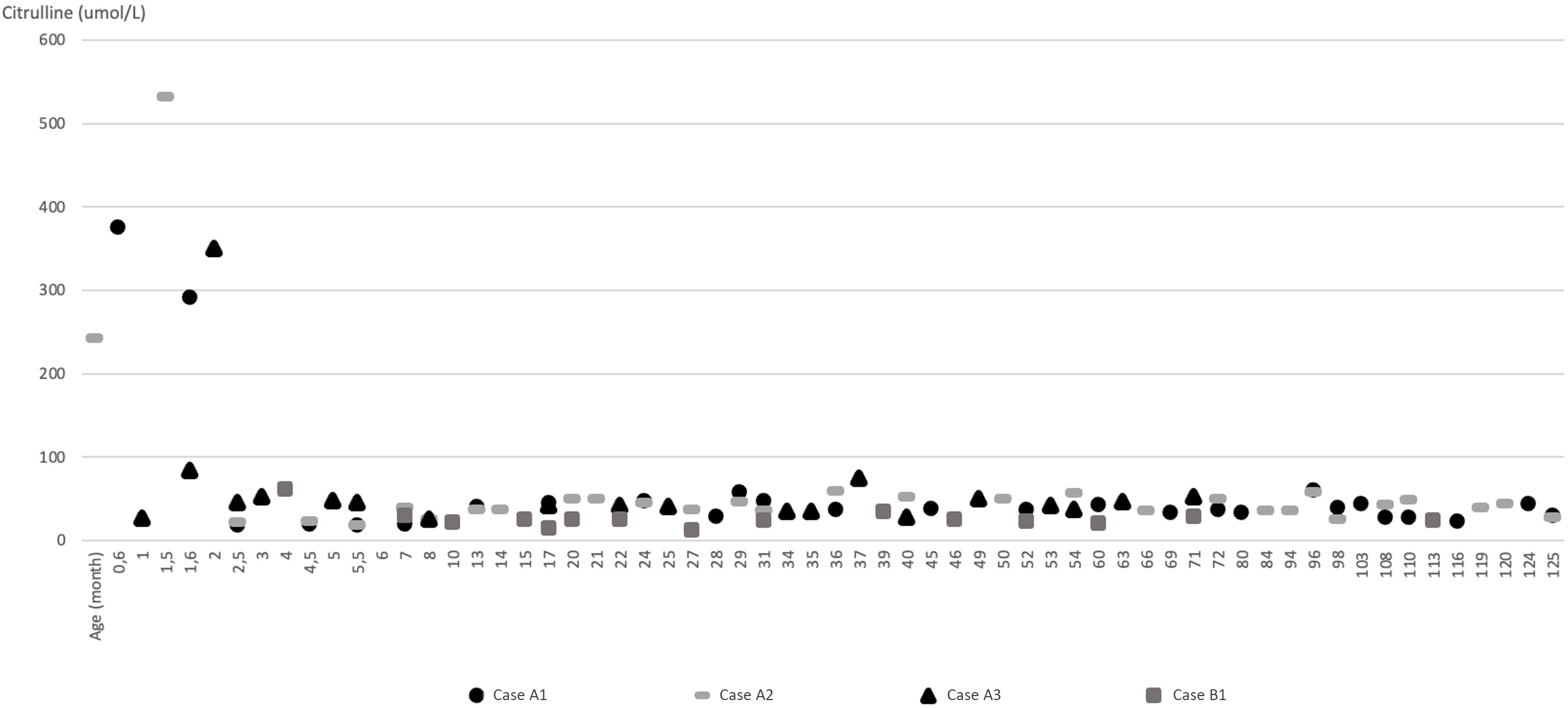
The patient was the first child of healthy non-consanguineous Turkish parents. The pregnancy was unremarkable. He was delivered at term via normal spontaneous delivery without asphyxia, with a birth weight of 3200 g (-0.3 SD), a length of 48 cm (-1.1 SD), and a head circumference of 33 cm (-1.5 SD). At the age of three and a half months, jaundice was observed. He was found to have cholestasis and a reducing substance was detected in his urine and was referred to our department at the age of five months. On physical examination, he weighed 6860 g (-0.68 SD) and measured 64 cm (-0.64 SD) in length. His liver was palpable 4 cm below the costal margin with no signs of splenomegaly. Laboratory evaluation showed cholestasis and mildly elevated liver transaminases (Table I). He also displayed increased AFP (3416 ng/mL; normal: <13.6). Acylcarnitines by tandem MS exhibited a free carnitine (C0) level of 56.5 µmol/L (RR: 8.6-90), tetradecanoyl carnitine (C14) of 0.99 µmol/L (RR: 0-0.8), linolenoyl carnitine (C18:2) of 1.25 µmol/L (RR: 0-0.9), and oleyl carnitine (C18:1) of 3.43 µmol/L (RR: 0-2.8). Bile acid analysis from a dried blood spot sample revealed elevated bile acids as indicative of cholestasis or liver disease (taurochenodeoxycholic acid [TCDC] 23.1 µmol/L [RR: <4 µmol/L], glycochenodeoxycholic acid [GCDC] 25.6 µmol/L [RR: <4 µmol/L], taurocholic acid [TC] 11.2 µmol/L [RR: <4 µmol/L] and glycocholic acid [GCA] 6.5 µmol/L [RR: <4 µmol/L]). Evaluation of urinary succinylacetone to screen for tyrosinemia type I was unremarkable. Plasma amino acid analysis displayed elevations in the concentrations of Cit, Thr, Met, tyrosine (Tyr), and Arg (Table II). Abdominal USG indicated normal liver echogenicity and a simple cyst was observed in the right kidney cortex. The ophthalmologic examination was normal. Genetic analyses revealed heterozygosity for a previously described variant c.74C>A (p.Ala25Glu) on exon 3 and a novel c.1359G>T (p.Lys453Asn) on exon 14 on the SLC25A13 gene.5 Parental Sanger sequencing confirmed that the c.74C>A (p.Ala25Glu) variant was maternally inherited, whereas the c.1359G>T (p.Lys453Asn) variant was paternally inherited, supporting a compound heterozygous state in the patient. MCT oil was administered with a galactose-free formula at the age of five months. His clinical condition and pathologic laboratory findings improved and diet was discontinued at 18 months of age when all abnormal laboratory findings had normalized. The biochemical parameters, including liver enzymes and synthetic functions, lipid profiles, carnitine and Cit levels, and ammonia remained within normal range, and he attained normal neurocognitive development at follow-up. Fig. 1 illustrates the plasma Cit values of all four patients during the follow-up period. At the latest outpatient visit at the age of 10 years, he weighed 40 kg (1.1 SD) and was 146 cm (1.42 SD) tall. Physical examination findings, abdominal USG, and blood tests including liver function tests were normal. The eye examination was completely normal.
Discussion
Here we describe four patients from two unrelated families from Türkiye; all of them had the NICCD phenotype and biallelic variants in the SLC25A13 gene. The patients were followed for periods of 11, 11, 10, and 7 years at our center. Due to the rarity of CD deficiency in non-Asian countries, the understanding of its clinical spectrum is still evolving, encompassing both infantile and late-onset forms.
All our patients presented with neonatal cholestasis, hypertransaminasemia, galactosuria and elevated Cit levels. However, there were notable variations in the severity of hyperbilirubinemia, with total bilirubin levels ranging from 5.2 mg/dL to 8.9 mg/dL in our cases, whereas previously published cases showed a broader range, with some cases exceeding 21 mg/dL.6 Direct bilirubin levels in our cohort were relatively mild (1.3–3.5 mg/dL), while published cases exhibited more severe cholestasis, reaching up to 12.3 mg/dL.6 AFP levels also displayed significant variability. While our cases exhibited moderately elevated AFP (3,416–121,000 ng/mL), other reported cases had extreme elevations, with some exceeding 450,000 ng/mL.6 Hyperammonemia and hypoglycemia were not observed in our cases. Despite the biochemical abnormalities, all patients showed favorable long-term outcomes following dietary intervention.
The patients with NICCD typically exhibit a mild increase in galactose and galactose-1-phosphate levels without a pronounced elevation in galactose-1-phosphate and UDP-galactose, as observed in epimerase deficiency.4 It is assumed that the inhibition of UDP-galactose epimerase by NADH, a competitor of NAD+ bound to the enzyme, may be the cause of secondary galactosemia in NICCD. Markers of galactosemia such as galactose, galactitol, and galactonate are frequently detected in urine samples of NICCD cases. The accumulation of galactitol may be implicated in the pathogenesis of clinical manifestations including jaundice, hepatosplenomegaly, hepatocellular insufficiency, and cataracts.7 All of our patients had elevated galactose levels at the time of diagnosis. We determined urinary-reducing substances while monitoring our patients instead of galactose levels as it was more easily accessible. With a galactose-restricted diet, urinary galactose excretion normalized. Despite negative results in patients during follow-up, one patient developed cataracts. Tazawa et al.8 reported that hypergalactosemia was detected in 6 out of 9 patients with NICDD, and 4 of these patients had also developed cataracts. Dimmock et al.5 reported a case who presented with an elevated Phe and total galactose the upper limit of normal on newborn screening with positive reducing substances in the urine. They reported detecting arginosuccinate synthetase (ASS) enzyme deficiency on skin fibroblast culture in NICDD patients.
In the reported cases from our country, patients presented with symptoms consistent with the literature, such as jaundice, hepatomegaly, and FTT (Table I). Clinically, our cases did not develop hepatic failure, and all had favorable long-term outcomes with dietary management. In contrast, some published cases demonstrated progressive hepatic dysfunction, with one requiring liver transplantation.2 Except for one patient who passed away shortly after the diagnosis, the other patients have remained asymptomatic. The deceased patient exhibited direct hyperbilirubinemia, a 20-fold elevation in liver transaminases, prolonged coagulation, and hypoglycemia. A reductant agent was detected in the urine, and galactose levels were found to be elevated. Despite the administration of MCT supplementation and a galactose-free formula, the patient succumbed to a sepsis attack during the observation phase.6
In CD, diet therapy is the only treatment option, if patients do not adhere to the lifelong diet, it may lead to a series of complications, such as cataracts, FTT, dyslipidemia, liver failure, and hyperammonemic encephalopathy.4 Patient 2 was advised to discontinue the dietary treatment, and the development of cataracts was observed during follow-up. While this finding does not necessarily predict the occurrence of other clinical manifestations, it may serve as an early indicator of disease progression and warrant closer monitoring of metabolic and hepatic parameters. The mechanisms leading to this transition have yet to be defined as these findings attributed to carbohydrate toxicity serve as predictive markers that can help to anticipate the initiation of CTLN2. In contrast, a high-protein/high-fat/low-carbohydrate diet prevents the onset of CTLN2. Some patients exhibit specific clinical features such as growth retardation, fatigue, weight loss, fatty liver, hyperlipidemia, hyperammonemia, and citrullinemia shortly before the onset of CTLN2. It is crucial to monitor patients for these pre-CTLN2 symptoms.4 External factors such as sugar intake, alcohol consumption, and specific medications like acetaminophen, as well as high-sugar solutions and/or glycerol, and surgical procedures, can aggravate these manifestations.9 Care should be provided especially to NICDD patients for the onset of CTLN2 with weight loss, height stagnation, easy fatigability, and high blood levels of ammonia, Cit, Thr/Ser ratio, and pancreatic secretory trypsin inhibitor (PSTI).4 Additionally, we recommend evaluating patients for galactose levels and cataracts.
To our knowledge, a total of four Turkish patients with CTLN2 have been reported in the literature to date. Köse et al.6 first reported three siblings with CTLN2 carrying the homozygous c.1478 G>A (p.Asp493Gly) variant in the SLC25A13 gene. The proband, a 15-year-old male, presented with intellectual disability, generalized tonic-clonic convulsions, and autism. He experienced delirium, and laboratory data showed hyperammonemia and elevated Cit levels. Ateş et al.10 reported a 30-year-old male patient with CTLN2 who developed hyperammonemic encephalopathy induced by sodium valproate. His blood ammonia level was normal at admission, but it increased to 339 µmol/l (RR: 9-97 µmol/l) after the initiation of multiple antiepileptic drugs due to seizures. Plasma amino acid analysis revealed moderately elevated levels of Cit (142.2 µmol/L [normal range: 17–46 µmol/L]) and Thr (312 µmol/L [normal range: 60–200 µmol/L]), with an increased Thr/Ser ratio. Molecular analysis revealed a homozygous c.848 G>T (p.Gly283Val) variant in the SLC25A13 gene. There have been no reported cases with the FTTDCD phenotype to date in our population.
The nonsense c.955C>T (p.Arg319*) variant in the SLC25A13 gene was first identified in Chinese NICCD patients and subsequently reported in a Japanese NICCD patient. This variant, located in exon 10, introduces a premature stop codon, predicted to lead to the early termination of the citrin protein.11,12 This variant occurs between the linker loop domain and the carrier domain, truncating the protein before it can form the full carrier domain required for mitochondrial transport. As this domain is essential for aspartate-glutamate transport, the mutation likely leads to a complete loss of citrin function (https://www.uniprot.org/). According to the American College of Medical Genetics and Genomics (ACMG) criteria, this variant is classified as pathogenic.13 The c.74C>A (p.Ala25Glu) variant in the SLC25A13 gene was first identified in an NICCD patient of Arabic descent. This variant, located in the N-terminal domain, predicted not to affect the transport activity of citrin, but it is expected to impair calcium regulation.5 Based on available data, this variant is classified as likely pathogenic according to ACMG criteria.13 There is also a Caucasian case presenting with the NICCD phenotype, who was compound heterozygous for a missense variant, c.74C>A (p.Ala25Glu), which produces an Ala-to-Glu substitution at position 25, and a nonsense variant, c.1081C>T (p.Arg361*).14 These publications have shown that the same variants are observed in different ethnic groups.
The c.1359G>T (p.Lys453Asn) variant has not been previously reported in the literature and was identified as very rare (0.0008%) in the gnomAD population. According to the ACMG criteria, this variant has received scores for PM2 and PP3 and was determined as a variant of uncertain significance (VUS). The functional effect was predicted to be disease-causing by MutationTaster with a score of 1, deleterious by Sorting Intolerant From Tolerant (SIFT) with a score of 0.00 (<0.05 is predicted to be deleterious), and harmful by PolyPhen-2 HumVar with a score of 1 (1.0 is predicted to be deleterious).13
A recent report identified a novel c.1610_1612delinsAT (p.Leu537TyrfsTer2) variant in an Indian patient, presenting with recurrent encephalopathy and hyperammonemia, underscoring this mutation’s potential to induce severe neuropsychiatric manifestations. The study also analyzed 79 citrin deficiency cases from 24 studies to establish genotype-phenotype correlations. The c.851_854del4 (p.Met285Profs*2) mutation demonstrated positive associations with alpha-fetoprotein, ammonia, and Tyr levels, while showing an inverse correlation with Thr. The IVS16ins3kb mutation was linked to elevated total and conjugated bilirubin, along with increased aspartate transaminase, whereas Cit levels were lower in these patients. Additionally, c.674C>A (p.Ser225*) and c.1645C>T (p.Gln549*) mutations were associated with a milder hepatic phenotype, suggesting a potential role of these mutations in modulating the severity of liver impairment in CD.15
A review of 138 Chinese NICCD patients revealed that homozygous mutations of 1638_1660dup, IVS6+5G>A, and IVS16ins3kb were exclusively observed in the acute liver failure (ALF) and liver dysfunction groups, suggesting a strong association with severe liver impairment. The high-frequency c.851_854delGTAT (p.Met285Profs*2) mutation was detected across acute liver failure, liver dysfunction, and non-liver dysfunction groups, indicating that this mutation may contribute to varying degrees of hepatic injury.16
The initial biochemical findings of NICCD may overlap with PKU or galactosemia, making early diagnosis challenging. In NICCD, an elevation in phenylalanine levels can be observed due to liver damage. However, unlike PKU, not only phenylalanine but also other amino acids that increase in liver dysfunction are elevated. Additionally, similar to galactosemia, galactosuria and the presence of reducing sugars in urine may suggest an impairment in galactose metabolism. However, classical galactosemia testing does not reveal a significant enzyme deficiency. Therefore, expanded metabolic screening and genetic analysis are crucial for distinguishing NICCD from other metabolic disorders.
According to our latest information, very few patients have been reported from Türkiye so far.6,17-20 The diagnosis of some patients may have been missed. Newborn screening for CD based on Cit level measurement by tandem MS has low sensitivity. Therefore, Kido et al. proposed that a new scoring system based on tandem MS, including levels of Arg, Cit, isoleucine+leucine, Tyr, and the C0 to glutarylcarnitine (C5-DC) ratio, was more sensitive.3 Implementing this enhanced screening method in our country could facilitate the diagnosis of new cases through newborn screening programs.
This study reported the diagnosis and the clinical course in four new genetically confirmed cases of NICCD patients of non-Asian origin. Information on the variant spectrum of SLC25A13 in the Turkish population is limited. The findings in this paper further expanded the genotypic spectrum and genotype-phenotype correlations of CD. Although cases are presenting with cataracts in cases of CD, a case of cataract recurrence during follow-up has not been reported.7,8 Therefore, it is important to follow patients throughout childhood, adolescence, and adulthood, including eye examination.
Ethical approval
Informed consent was obtained from all individuals included in this study, or their parents / legal guardians.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Palmieri F, Scarcia P, Monné M. Diseases caused by mutations in mitochondrial carrier genes SLC25: a review. Biomolecules 2020; 10: 655. https://doi.org/10.3390/biom10040655
- Kido J, Häberle J, Sugawara K, et al. Clinical manifestation and long-term outcome of citrin deficiency: report from a nationwide study in Japan. J Inherit Metab Dis 2022; 45: 431-444. https://doi.org/10.1002/jimd.12483
- Kido J, Häberle J, Tanaka T, et al. Improved sensitivity and specificity for citrin deficiency using selected amino acids and acylcarnitines in the newborn screening. J Inherit Metab Dis 2024; 47: 1134-1143. https://doi.org/10.1002/jimd.12673
- Okano Y, Ohura T, Sakamoto O, Inui A. Current treatment for citrin deficiency during NICCD and adaptation/compensation stages: strategy to prevent CTLN2. Mol Genet Metab 2019; 127: 175-183. https://doi.org/10.1016/j.ymgme.2019.06.004
- Dimmock D, Maranda B, Dionisi-Vici C, et al. Citrin deficiency, a perplexing global disorder. Mol Genet Metab 2009; 96: 44-49. https://doi.org/10.1016/j.ymgme.2008.10.007
- Köse MD, Kagnici M, Özdemir TR, et al. Clinical findings in five Turkish patients with citrin deficiency and identification of a novel mutation on SLC25A13. J Pediatr Endocrinol Metab 2020; 33: 157-163. https://doi.org/10.1515/jpem-2019-0377
- Saheki T, Kobayashi K, Iijima M, et al. Adult-onset type II citrullinemia and idiopathic neonatal hepatitis caused by citrin deficiency: involvement of the aspartate glutamate carrier for urea synthesis and maintenance of the urea cycle. Mol Genet Metab 2004; 81(Suppl 1): 20-26. https://doi.org/10.1016/j.ymgme.2004.01.006
- Tazawa Y, Kobayashi K, Abukawa D, et al. Clinical heterogeneity of neonatal intrahepatic cholestasis caused by citrin deficiency: case reports from 16 patients. Mol Genet Metab 2004; 83: 213-219. https://doi.org/10.1016/j.ymgme.2004.06.018
- Kido J, Makris G, Santra S, Häberle J. Clinical landscape of citrin deficiency: a global perspective on a multifaceted condition. J Inherit Metab Dis 2024; 47: 1144-1156. https://doi.org/10.1002/jimd.12722
- Ateş BE, Demir T, Kor D, et al. Sodium valproate-induced adult-onset type 2 citrullinemia. Turkish Journal of Neurology 2022; 28: 197-199. https://doi.org/10.4274/tnd.2022.37539
- Song YZ, Li BX, Chen FP, et al. Neonatal intrahepatic cholestasis caused by citrin deficiency: clinical and laboratory investigation of 13 subjects in mainland of China. Dig Liver Dis 2009; 41: 683-689. https://doi.org/10.1016/j.dld.2008.11.014
- Song YZ, Sheng JS, Ushikai M, et al. Identification and diagnosis of three novel mutations in SLC25A13 gene of neonatal intrahepatic cholestasis caused by citrin deficiency. Zhonghua Er Ke Za Zhi 2008; 46: 411-415.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405-424. https://doi.org/10.1038/gim.2015.30
- Avdjieva-Tzavella DM, Ivanova MB, Todorov TP, et al. First Bulgarian case of citrin deficiency caused by one novel and one recurrent mutation in the SLC25A13 gene. Genet Couns 2014; 25: 271-276.
- Radha Rama Devi A, Naushad SM. SLC25A13 c.1610_1612delinsAT mutation in an Indian patient and literature review of 79 cases of citrin deficiency for genotype-phenotype associations. Gene 2018; 668: 190-195. https://doi.org/10.1016/j.gene.2018.05.076
- Jiang M, Peng M, Lu Z, et al. Features of liver injury in 138 Chinese patients with NICCD. J Pediatr Endocrinol Metab 2023; 36: 1154-1160. https://doi.org/10.1515/jpem-2023-0026
- Zeybek AC, Kiykim E, Zubarioglu T, et al. Citrin deficiency: an infant incidentally detected by Phenylketonuria screening with a novel mutation in SLC25A13 gene. Genet Couns 2015; 26: 409-413.
- Şeker-Yılmaz B, Kör D, Tümgör G, Ceylaner S, Önenli-Mungan N. p.Val452Ile mutation of the SLC25A13 gene in a Turkish patient with citrin deficiency. Turk J Pediatr 2017; 59: 311-314. https://doi.org/10.24953/turkjped.2017.03.012
- Bilgin H, Bilge S, Binici M, Tekes S. Clinical, biochemical, and genotypical characteristics in urea cycle mitochondrial transporter disorders. Eur Rev Med Pharmacol Sci 2024; 28: 1873-1880. https://doi.org/10.26355/eurrev_202403_35601
- Saritaş Nakip Ö, Yıldız Y, Tokatlı A. Retrospective evaluation of 85 patients with urea cycle disorders: one center experience, three new mutations. J Pediatr Endocrinol Metab 2020; 33: 721-728. https://doi.org/10.1515/jpem-2019-0413
Copyright and license
Copyright © 2025 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









