
Abstract
Background. Congenital tufting enteropathy (CTE) is a rare autosomal recessive enteropathy characterized by intractable watery diarrhea independent of feeding, electrolyte imbalances and severe malnutrition. The aim of this case series is to highlight the characteristic yet easily overlooked histological features of tufting enteropathy, especially in cases where initial biopsies may be reported as nonspecific, and to emphasize the importance of clinicopathological correlation and genetic confirmation.
Case Presentation. Here we report 3 patients with tufting enteropathy who have the characteristic histological findings and demonstrate the use of EpCAM (epithelial cell adhesion molecule) immunohistochemistry. One case was diagnosed by histopathological examination, while the other 2 were diagnosed by genetic analysis.
Conclusions. These cases illustrate that the histopathological features of CTE, though subtle, are usually present from the outset and can be recognized when specifically sought. Correlation with genetic testing is essential for confirmation, but increased awareness of the characteristic epithelial tufts may reduce diagnostic delay and improve patient management.
Keywords: congenital diarrhea, EpCAM, intestinal epithelial dysplasia, pediatric, tufting enteropathy
| IEL: intraepithelial lymphocyte, N/A: not available. | ||||||
| Table I. Summary of clinical features, genes and histologic findings reported in literature | ||||||
| Study |
|
|
Symptoms | Genetic variants | Histological findings | Outcome |
| Reifen et al1 |
|
|
Diarrhea, vomiting | N/A* | N/A | 1 died |
| Patey et al4 |
|
|
Diarrhea | N/A | Villous atrophy, focal epithelial tufts, decreased number of IELs | Unknown |
| Goulet et al10 |
|
|
Diarrhea | N/A | Villous atrophy, epithelial tufting | Unknown |
| Bird et al5 |
|
|
Diarrhea, choanal atresia; eye, hematologic and hair abnormalities | N/A | Villous atrophy, crypt hyperplasia, chronic inflammation, tufting of surface enterocytes | Unknown |
| Sivagnanam et al11 |
|
|
Diarrhea | EPCAM in 4/5 cases | Crowding of epithelial cells and tufting | Unknown |
| Sivagnanam et al6 |
|
|
Diarrhea, cholestatic liver disease | SPINT2 | N/A | Alive |
| Salomon et al14 |
|
|
Diarrhea, superficial punctuated keratitis, choanal atresia, other atresia, dermatological anomalies, and bone malformations | EPCAM in 41 cases, SPINT2 in 12 cases | Villous atrophy, crowding epithelium/tufts, abnormal crypts | 8 died |
| Ranganathan et al7 |
|
|
Diarrhea | N/A | Focal epithelial tufts and vacuolation | Alive |
| Pathak et al12 |
|
|
Diarrhea | EPCAM | N/A | Alive |
| Holt-Danborg et al8 |
|
|
Diarrhea, Choanal atresia, enterocutaneous fistula, atrial septal defect, cleft lip and palate, and toe abnormalities | SPINT2 | Partial villous atrophy, normal brush border, focal epithelial tufts | Alive |
| Ayyildiz Civan et al9 |
|
|
Diarrhea | EPCAM | Total villous atrophy, mild inflammation | 3 died |
| Ozler et al19 |
|
|
Diarrhea, tenosynovitis | EPCAM | Partial villous atrophy, crypt hyperplasia, focal tufting in surface epithelium, normal number of IELs | Alive (after long term follow up) |
Introduction
Congenital tufting enteropathy (CTE) was first described in 19941 and its incidence is estimated at 1/50,000-100,000 live births in Western Europe.2 For this rare disease, approximately 200 cases have been reported (details are summarized in Table I).3-10 Biallelic inactivation of epithelial cell adhesion molecule gene (EPCAM), which is located on chromosome 2p21, was identified as the cause of CTE in 2008.11 More recently, variants in SPINT2 (serine peptidase inhibitor kunitz-type 2) have also been implicated in the syndromic form of this disease, which causes an indirect loss of epithelial cell adhesion molecule (EpCAM) protein due to proteolysis by activation of matriptase.12 Of all the cases reported in English literature, approximately 74% have EPCAM variants, while 26% show variants in SPINT2.13
Variants in or loss of EPCAM lead to many defects, the most important being the disruption of the cell-cell junction which leads to defective barrier function. Disruption of the intestinal epithelial homeostasis, defective enterocyte function and impaired cell-matrix interactions have also been found in CTE patients.3 Clinically, these patients usually present in the first few months of life with intractable diarrhea and nutrient malabsorption, resulting in impaired growth.11 In addition to primary intestinal symptoms, patients, especially those who have the syndromic form associated with SPINT2 variants13,14 can have extra-intestinal symptoms, such as superficial punctuated keratitis, corneal erosions15, cataracts16, skeletal dysplasia, cholestatic liver disease, chronic arthritis, bone and dermatological abnormalities.3 Typical histopathological findings of CTE are partial or total villous atrophy, crypt hyperplasia without inflammation and focal epithelial tufts. The tufts consist of tightly packed enterocytes at villous tips and basement membrane abnormalities which results in teardrop-like configurations.3 However, cases reported in the literature are clinically heterogenous, with varying severity and not all cases display the typical histopathological findings, which poses a diagnostic challenge.17 BerEP4 immunohistochemistry (monoclonal antibody to EpCAM) can be helpful in the diagnosis of cases that do not have the typical morphological findings. In addition to intestinal epithelial cells, loss of BerEP4 can also be seen in hepatocytes and bile duct epithelium in liver biopsies of patients with tufting enteropathy.18
Although large series of tufting enteropathy have been published, most emphasize the genetic basis or clinical spectrum.14-16 Our series draws attention to the diagnostic challenge that these cases pose and highlights the fact that the characteristic histological findings of tufting enteropathy can usually be detected if carefully sought, which may shorten diagnostic delay.
Case Presentations
Case 1
A 2-month-old girl was admitted to the pediatric intensive care unit with prerenal acute kidney failure due to profuse watery diarrhea that had been resistant to treatment. The family pedigree showed that the parents were consanguineous (second-degree cousins). There were two family members reported to have had similar symptoms during childhood, including the father’s younger brother who died in early infancy. Unfortunately, no reliable information on their age, sex, clinical features, or outcome was available. The patient had symptoms of watery diarrhea that occurred 8 to 9 times a day. Before admission to our center, she was hospitalized for two weeks at another hospital and had received antibiotic treatment. Breastfeeding was discontinued due to a preliminary diagnosis of cow’s milk protein allergy. The patient was fed with an enteral nutrition formula however oral intake was completely discontinued after a few days due to worsening of symptoms.
On admission, the patient was found to be severely dehydrated. The birth weight of the patient was 2900 grams (-0.8 standard deviation score [SDS]). Her body weight at admission was 2600 grams (-7.2 SDS) consistent with severe growth faltering. Laboratory examination revealed metabolic acidosis with a blood pH of 7.16 , bicarbonate of 9 mEq/L and creatinine of 1.52 mg/dL (normal range, 0.2-0.4 mg/dL). Stool examination revealed no reducing substances, parasites, or amoebae. After starting intravenous fluids and stabilization, endoscopic biopsies from various parts of the stomach, small intestine, and colon were taken to shed light on the etiology of the chronic diarrhea. The preliminary diagnoses included cytomegalovirus (CMV) colitis, enteric anendocrinosis, lymphangiectasia and cow’s milk protein allergy.
At initial examination, the endoscopic biopsies showed no specific findings. The diagnoses of lymphangiectasia and cow’s milk protein allergy were ruled out immediately because there were no dilated lymphatic channels or an increase in eosinophils in the lamina propria. CMV and chromogranin immunohistochemistry studies were performed for the remaining diagnoses. CMV was negative in all tissues. Chromogranin revealed fewer neuroendocrine cells (1-2 NECs per crypt) than the normal number of 4-5 NECs per crypt. However, this was not enough to support the diagnosis of enteric anendocrinosis as most cases lack NECs completely or have 1 NEC per 10 crypts. After these findings, whole exome sequencing (WES) was performed to evaluate variants in NEUROG3. However, the WES study revealed a pathogenic homozygous EPCAM variant (NM_002354.3: c.227C>G [p.Ser76Ter]) instead. The biopsies were re-evaluated in this view with additional hematoxylin and eosin (H&E) staining and immunohistochemical staining for EpCAM. In the H&E sections of the duodenum, terminal ileum and colon, focal, subtle epithelial disorganization and tear-drop formation were found (Fig. 1A-B). Immunohistochemistry revealed complete loss of EpCAM expression in the intestinal and colonic tissues, which is specific for CTE (Fig. 1C).
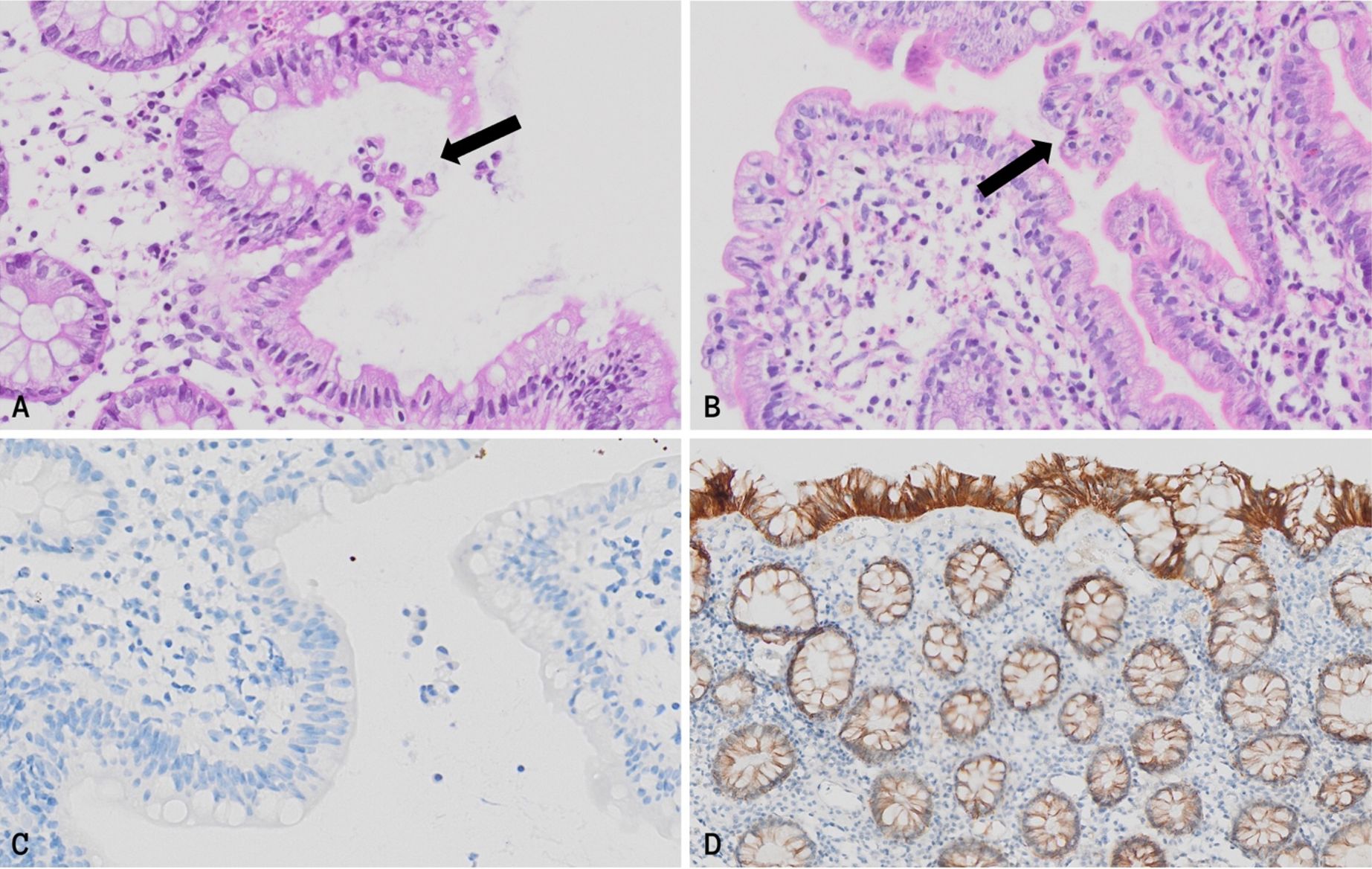
The patient was fed with total parenteral nutrition after the diagnosis and per oral feeding could not be achieved. At 1 year of age, despite parenteral nutrition support, the patient developed progressive feeding intolerance with vomiting and abdominal distension, followed by respiratory distress and circulatory collapse. Cardiopulmonary resuscitation was unsuccessful, and the patient could not be resuscitated. The outcome was attributed to complications of severe malnutrition and intestinal failure.
Case 2
A 17-month-old boy was admitted to the intensive care unit with severe malnutrition and dehydration due to watery diarrhea that had been increasing since birth. The parents were first cousins and were healthy. They had another child who passed away soon after birth due to similar symptoms. The patient was born at 35 weeks of gestation, weighing 2700 grams. When he was admitted to our center at 17 months of age, he weighed 3,600 g (-9.7 SDS) and his height was 61 cm (-7.7 SDS). The patient was previously hospitalized in multiple centers and no etiology could be identified for the intractable diarrhea. Endoscopic and colonoscopic biopsies were performed and they revealed crypt hyperplasia, villous atrophy, and an increase in intraepithelial lymphocytes. Soon after admission the clinical condition of the patient deteriorated, and he passed away. The family consented to an autopsy and molecular genetic studies. The autopsy revealed villous atrophy in the small intestine, hydrops in the gallbladder, severe malnutrition and cachexia and reactive enlargement of mesenteric lymph nodes. The duodenum sections were examined by electron microscopy. There were a few microvilli however no microvillus inclusions were identified. No definitive cause could be identified for chronic diarrhea. To rule out genetic causes, whole exome sequencing was performed. A homozygous missense variant in EPCAM (NM_002354.3: c.757G>A [p.Asp253Asn]) compatible with the disease phenotype in the affected individual was found. This case has been previously published in 2022 by Güvenoğlu et al.17 The additional details regarding genetic studies can be found in their article. We reviewed the autopsy sections of this patient and performed EpCAM immunohistochemistry on the duodenal tissue. The histologic examination of the duodenum showed villous atrophy and classic teardrop formation on the surface (Fig. 2A). In addition, there was a slight increase in intraepithelial lymphocytes. The BerEP4 immunohistochemistry was negative as expected (Fig. 2B).
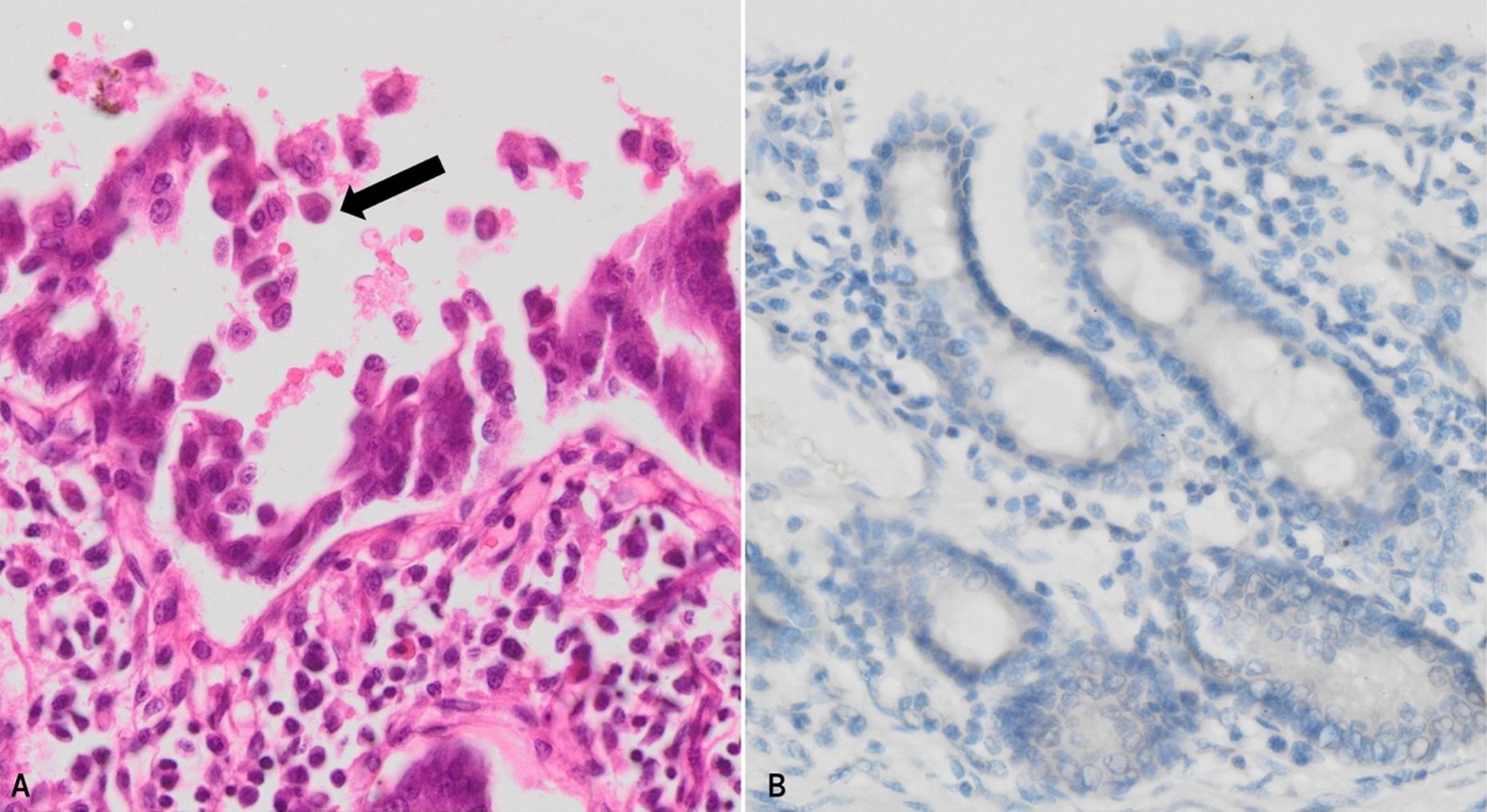
Case 3
The third case is a 10-month-old male infant who had vomiting and diarrhea that commenced 2 days following birth. The parents were first-degree relatives who were healthy. The patient was initially examined for metabolic diseases due to the presence of reducing substances in urine. However, tandem mass spectrometry was normal. Sweat test for cystic fibrosis was also negative. The endoscopic biopsy from the duodenum was diagnostic as it revealed clustering of epithelial cells forming characteristic teardrop cells (Fig. 3A). The brush border was examined with periodic acid-Schiff (PAS) staining and was preserved. There was also partial villous atrophy, a minimal increase in lymphocytes in the lamina propria and crypt hyperplasia. The EpCAM immunohistochemistry was negative (Fig. 3B). Genetic testing could not be performed for this patient and no follow-up information was available.
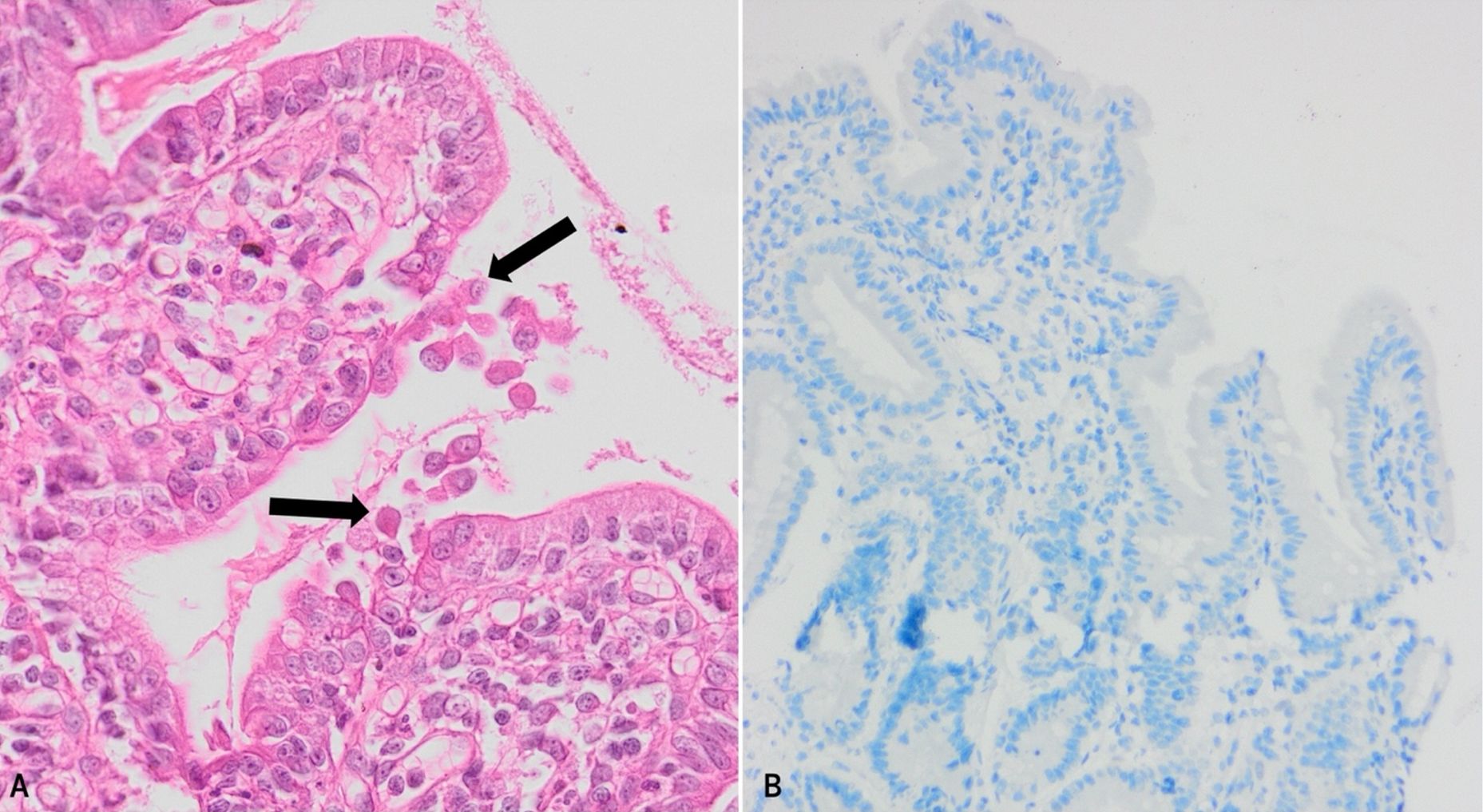
Informed consent could not be obtained from the families because one case dated back more than 10 years and the other two families could not be reached. Therefore, permission from the ethics committee was obtained (number: 2024/17-03).
Discussion
Our three cases illustrate the diagnostic challenges of CTE. Although the disease is genetically defined, the histological findings can also aid in the diagnosis. In this section, we highlight the variability of the clinical course and emphasize the complementary value of histopathology and genetics in diagnosis.
While most patients depend on total parenteral nutrition (PN), there are reported cases where weaning from PN was achieved.11,19 One of these case reports describing two siblings with tufting enteropathy showed complete loss of EpCAM in immunohistochemistry and a homozygous variant in the EPCAM gene. Certain patients may have a better prognosis due to unknown factors.19 The only accepted treatment to date is small bowel transplantation, which has many complications such as liver failure, infections and rejection.3
Although the diagnosis of tufting enteropathy usually depends on genetic analysis, the histopathological examination can also be diagnostic if the pathologist is aware of CTE in the differential diagnosis. However, the histological findings of CTE can be overlooked and interpreted as nonspecific, which was the major challenge in 2 of our cases. Only after genetic confirmation did retrospective review reveal the subtle epithelial tufts and teardrop formations. This highlights the risk of underdiagnosis if CTE is not specifically considered. When provided with a history of intractable diarrhea in an infant, the gastrointestinal biopsies should be carefully examined for epithelial tufts and villous atrophy, and BerEP4 immunohistochemistry can be performed on cases showing villous atrophy without inflammation or when the histological findings are equivocal. Genetic testing remains essential for definitive diagnosis, prognosis, and family counseling, but increased histopathological awareness can reduce diagnostic delay and guide appropriate workup.14
In conclusion, our experience shows that while subtle, the histological features of CTE are characteristic when interpreted in the correct clinical context. Greater awareness of these findings, combined with timely EpCAM immunohistochemistry and genetic analysis, can improve recognition of this rare but important cause of intractable infantile diarrhea.
Ethical approval
The study was approved by Hacettepe University Ethics Committee (date: October 08, 2024, number: 2024/17-03).
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Reifen RM, Cutz E, Griffiths AM, Ngan BY, Sherman PM. Tufting enteropathy: a newly recognized clinicopathological entity associated with refractory diarrhea in infants. J Pediatr Gastroenterol Nutr 1994; 18: 379-385.
- Goulet O, Salomon J, Ruemmele F, de Serres NPM, Brousse N. Intestinal epithelial dysplasia (tufting enteropathy). Orphanet J Rare Dis 2007; 2: 20. https://doi.org/10.1186/1750-1172-2-20
- Das B, Sivagnanam M. Congenital tufting enteropathy: biology, pathogenesis and mechanisms. J Clin Med 2020; 10: 19. https://doi.org/10.3390/jcm10010019
- Patey N, Scoazec JY, Cuenod-Jabri B, et al. Distribution of cell adhesion molecules in infants with intestinal epithelial dysplasia (tufting enteropathy). Gastroenterology 1997; 113: 833-843. https://doi.org/10.1016/s0016-5085(97)70178-1
- Bird LM, Sivagnanam M, Taylor S, Newbury RO. A new syndrome of tufting enteropathy and choanal atresia, with ophthalmologic, hematologic and hair abnormalities. Clin Dysmorphol 2007; 16: 211-221. https://doi.org/10.1097/MCD.0b013e328274264b
- Sivagnanam M, Janecke AR, Müller T, Heinz-Erian P, Taylor S, Bird LM. Case of syndromic tufting enteropathy harbors SPINT2 mutation seen in congenital sodium diarrhea. Clin Dysmorphol 2010; 19: 48. https://doi.org/10.1097/MCD.0b013e328331de38
- Ranganathan S, Schmitt LA, Sindhi R. Tufting enteropathy revisited: the utility of MOC31 (EpCAM) immunohistochemistry in diagnosis. Am J Surg Pathol 2014; 38: 265-272. https://doi.org/10.1097/PAS.0000000000000106
- Holt-Danborg L, Vodopiutz J, Nonboe AW, et al. SPINT2 (HAI-2) missense variants identified in congenital sodium diarrhea/tufting enteropathy affect the ability of HAI-2 to inhibit prostasin but not matriptase. Hum Mol Genet 2019; 28: 828-841. https://doi.org/10.1093/hmg/ddy394
- Ayyıldız Civan H, Leitner C, Östreicher I, et al. Three novel EPCAM variants causing tufting enteropathy in three families. Children (Basel) 2021; 8: 503. https://doi.org/10.3390/children8060503
- Goulet OJ, Brousse N, Canioni D, Walker-Smith JA, Schmitz J, Phillips AD. Syndrome of intractable diarrhoea with persistent villous atrophy in early childhood: a clinicopathological survey of 47 cases. J Pediatr Gastroenterol Nutr 1998; 26: 151-161. https://doi.org/10.1097/00005176-199802000-00006
- Sivagnanam M, Mueller JL, Lee H, et al. Identification of EpCAM as the gene for congenital tufting enteropathy. Gastroenterology 2008; 135: 429-437. https://doi.org/10.1053/j.gastro.2008.05.036
- Pathak SJ, Mueller JL, Okamoto K, et al. EPCAM mutation update: variants associated with congenital tufting enteropathy and Lynch syndrome. Hum Mutat 2019; 40: 142-161. https://doi.org/10.1002/humu.23688
- Cai C, Chen Y, Chen X, Ji F. Tufting enteropathy: a review of clinical and histological presentation, etiology, management, and outcome. Gastroenterol Res Pract 2020; 2020: 5608069. https://doi.org/10.1155/2020/5608069
- Salomon J, Goulet O, Canioni D, et al. Genetic characterization of congenital tufting enteropathy: epcam associated phenotype and involvement of SPINT2 in the syndromic form. Hum Genet 2014; 133: 299-310. https://doi.org/10.1007/s00439-013-1380-6
- Heinz-Erian P, Müller T, Krabichler B, et al. Mutations in SPINT2 cause a syndromic form of congenital sodium diarrhea. Am J Hum Genet 2009; 84: 188-196. https://doi.org/10.1016/j.ajhg.2009.01.004
- Roche O, Putterman M, Salomon J, et al. Superficial punctate keratitis and conjunctival erosions associated with congenital tufting enteropathy. Am J Ophthalmol 2010; 150: 116-121.e1. https://doi.org/10.1016/j.ajo.2010.01.034
- Güvenoğlu M, Şimşek-Kiper PÖ, Koşukcu C, et al. Homozygous missense epithelial cell adhesion molecule variant in a patient with congenital tufting enteropathy and literature review. Pediatr Gastroenterol Hepatol Nutr 2022; 25: 441-452. https://doi.org/10.5223/pghn.2022.25.6.441
- Chen S, Goldsmith JD, Fawaz R, Al-Ibraheemi A, Perez-Atayde AR, Vargas SO. Liver pathology, including MOC31 immunohistochemistry, in congenital tufting enteropathy. Am J Surg Pathol 2021; 45: 1091-1097. https://doi.org/10.1097/PAS.0000000000001710
- Ozler O, Brunner-Véber A, Fatih P, Müller T, Janecke AR, Arikan C. Long-term follow-up of tufting enteropathy caused by EPCAM mutation p.Asp253Asn and absent EPCAM expression. JPGN Rep 2020; 2: e029. https://doi.org/10.1097/PG9.0000000000000029
Copyright and license
Copyright © 2026 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









