
Abstract
Background. Musculoskeletal manifestations of graft-versus-host disease (GVHD) are rare but often result in mobility impairments, reducing the patient’s quality of life. Typically, such diagnoses are made based on clinical findings without the need for performing a muscle biopsy.
Case Presentation. A 12-year-old boy diagnosed with acute myeloblastic leukemia (M2 subtype) underwent allogeneic hematopoietic stem cell transplantation (HSCT) due to a molecular relapse before his last chemotherapy cycle. Cyclosporine prophylaxis was stopped three months after transplantation, but the patient developed ocular, cutaneous, and oral chronic GVHD at four, five, and seven months after transplantation, respectively, for which intermittent steroid treatment and mycophenolate mofetil were given. All signs of GVHD resolved by one year after transplant, and immunosuppressive treatment was stopped; however, three months later, he experienced muscular weakness in bilateral upper and lower extremities. Subsequently, immunosuppressive treatment was restarted following a muscle biopsy.
Conclusion. Diagnosing musculoskeletal GVHD is challenging due to the lack of reliable parameters for histopathological diagnosis, and initial clinical findings can be mistaken for steroid-induced myopathy or inflammatory dermatomyopathies. We applied methylprednisolone, mycophenolate mofetil and extracorporeal photopheresis for treatment, and the clinical findings completely improved with these treatments.
Keywords: graft-versus-host disease, allogeneic bone marrow transplantation, musculoskeletal symptoms, inflammatory dermatomyopathy
Introduction
Graft-versus-host disease (GVHD) is an immune-mediated condition often encountered after allogeneic bone marrow transplantation when the donor’s immunocompetent T-cells deem the recipient’s antigens, known as human leukocyte antigen, as foreign. GVHD represents a significant issue for patients undergoing allogeneic hematopoietic stem cell transplantation (HSCT). Typically, acute GVHD (aGVHD) commonly affects the skin, liver, and gastrointestinal tract, while chronic GVHD (cGVHD) can affect nearly any organ system, with the gastrointestinal tract, oropharynx, skin, eyes, urogenital tract, lungs, and lymphohematopoietic system being the most frequently involved.1
Musculoskeletal GVHD is a rare manifestation of cGVHD with an incidence rate of 0.6% (49 cases per 1,000,000,000 people/year).1 It is extremely rare in children, with no prevalence data available, and only a few pediatric cases have been reported so far.2 However, the condition can be potentially life-threatening and causes significant disability, impairment, and a severe decline in the patient’s quality of life.2-11 This case report describes the histopathological findings of musculoskeletal GVHD in a pediatric patient with acute myeloblastic leukemia (M2 subtype; AML-M2).
Case presentation
A 12-year-old boy presented with eye swelling and pancytopenia. Bone marrow aspiration cytology showed 40% myeloblastic cells, and flow cytometry revealed CD13, CD33, CD117, and myeloperoxidase positivity. Further bone marrow genetic analysis revealed trisomy-8 and t(8;21) (AML-ETO-1) positivity on polymerase chain reaction (PCR) testing, confirming the diagnosis of AML-M2. Accordingly, the AML Berlin-Frankfurt-Münster (BFM) 2019 chemotherapy protocol was initiated. After induction treatment, initially, the AML-ETO-1 PCR turned negative but eventually returned to positive before the fifth and final chemotherapy cycles. Consequently, he was transferred to a stem cell transplantation unit. As no suitable donor was found within the family, HSCT was performed from a 10/10 matched unrelated donor following the FLAG-IDA (fludarabine, cytarabine, granulocyte colony stimulating factor - idarubicin) protocol. The following myeloablative conditioning regimen was included – busulfan (3.8 mg/kg/day, days -7 to -4; cyclophosphamide 60 mg/kg/day, days -3 to -2; and melphalan 140 mg/m2/day, last day before HSCT). Additionally, cyclosporine and methotrexate were used for GVHD prophylaxis.
Cyclosporine prophylaxis was stopped three months after transplantation. In the fourth month, there were signs of ocular GVHD, and at five months post-transplant, the patient developed grade III skin GVHD. Accordingly, mycophenolate mofetil and steroids were added to his regimen. At this time, he had 100% chimerism, and the bone marrow was in molecular remission. Although his skin and eye symptoms resolved, he developed oral grade II GVHD in the seventh month during the steroid tapering phase, necessitating an increase in the steroid dose.
By the end of the first year, the GVHD had resolved, and immunosuppressive therapies had been discontinued; muscle, joint, and neurological examinations remained normal during this period. However, three months after immunosuppressive therapy cessation, he exhibited muscular weakness in all four limbs, primarily in the lower limbs. Biochemical tests and blood counts, including creatine kinase (CK: 34 IU/L), were normal, and he tested negative for Epstein-Barr virus and cytomegalovirus PCR, Lyme disease serology, rheumatoid factor, antinuclear antibody, anti-dsDNA, anti-cardiolipin antibodies immunoglobulin G, and immunoglobulin M. The patient was uncooperative; hence, an electromyography (EMG) could not be performed, but nerve conduction tests were within normal limits. A muscle biopsy confirmed muscle involvement in cGVHD. Eventually, extracorporeal photopheresis was planned, and steroid (2 mg/kg/day) and mycophenolate mofetil (1500 mg/m2) were reintroduced. During the treatment, he developed a cough, which was diagnosed as pulmonary GVHD based on pulmonary function tests and thoracic computed tomography. All symptoms resolved after receiving two months of extracorporeal photopheresis.
Histopathological findings
Microscopic examination of the muscle biopsy specimen showed distortions in the diameter and shape of the muscle fibers. Scattered atrophic fiber structures were seen in the periphery of the fiber bundles, with no fiber grouping. However, the internal architecture of the muscle fibers exhibited normal nuclear retraction (< 3%), with no evidence of vacuoles or accumulations. There was no increase in the endomysial fibrous tissue, while the perimysial connective tissue appeared edematous and degenerated (Fig. 1). An inflammatory cellular reaction, comprising of macrophages, lymphocytes, and a few plasma cells, was predominant in the perimysial area and noticeable in the endomysial area, particularly around the capillary vessels (Fig. 2). Histochemical and enzyme staining revealed that the distribution and ratio of type I and type II fibers were preserved, and no abnormal glycogen or fat accumulation was detected. Cytochrome c oxidase (COX) and succinate dehydrogenase enzyme staining displayed no loss of enzymatic activity or mitochondrial aggregates. Immunohistochemical staining revealed sarcolemmal upregulation of major histocompatibility complex (MHC)-1, which was slightly more pronounced in the perifascicular fibers. Acid phosphatase and C5b9 staining exhibited characteristic features of inflammatory myopathy, with positive staining in macrophages and capillary structures (Fig. 3 and Fig. 4). Finally, staining of the cluster of differentiation 3 (CD3), CD4, CD8, CD20, and CD68 cells showed a cellular reaction rich in CD8-positive T cells and macrophages. When combined with the patient’s history, the histopathological findings were consistent with muscle involvement in cGVHD.
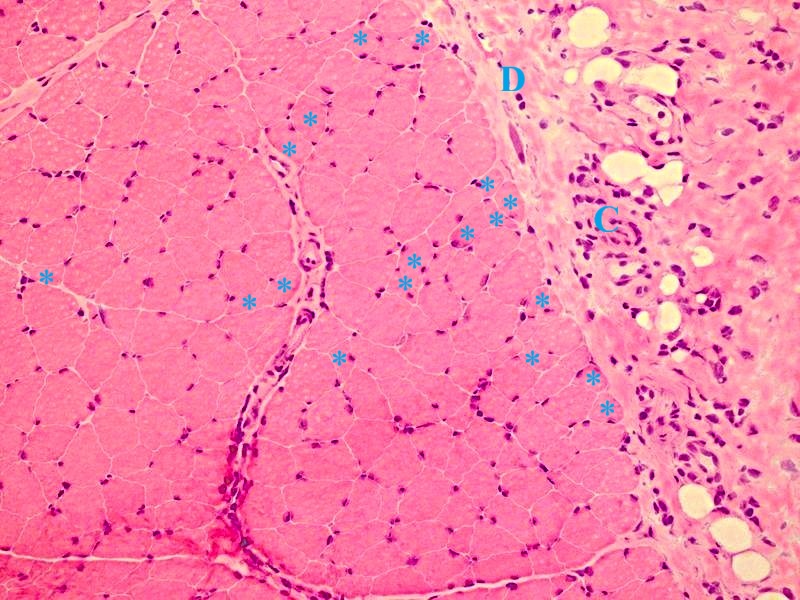
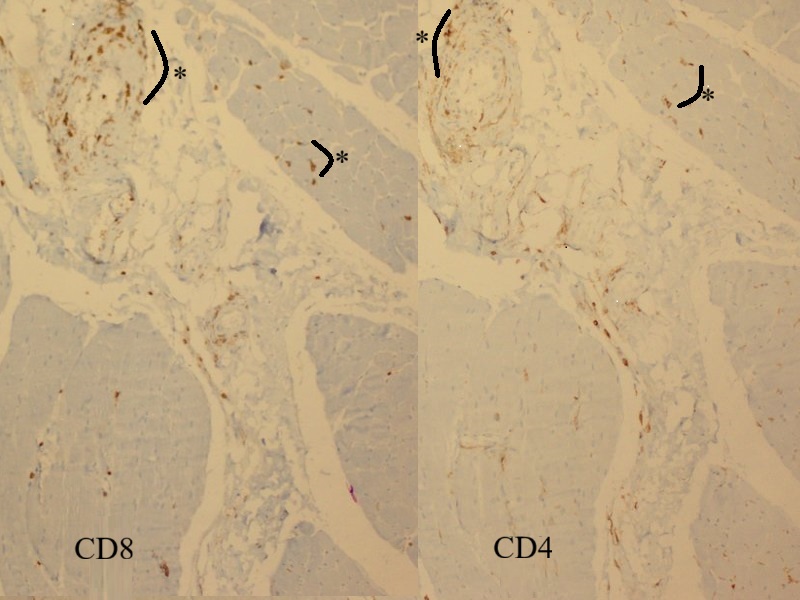
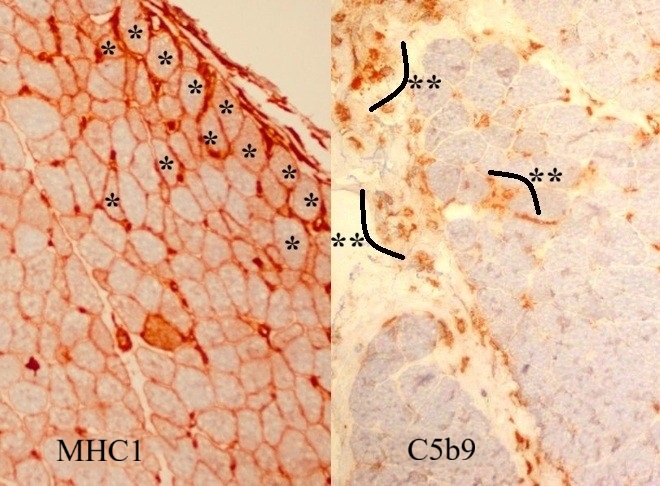
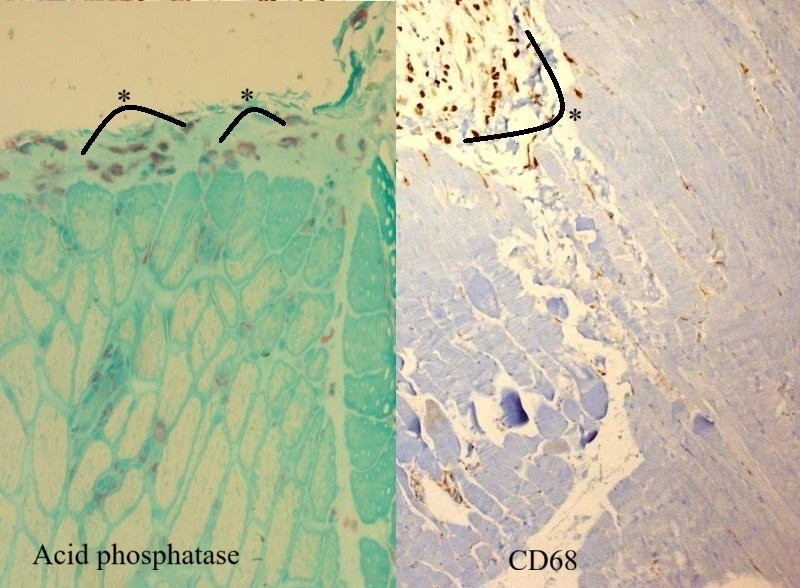
Informed consent was obtained from the patient’s parents, who agreed to the publication of the case findings.
Discussion
This case report underscores the importance of recognizing rare manifestations of cGVHD, particularly in pediatric patients. The existing literature affirms that musculoskeletal involvement in cGVHD implies a unique pathological process. In the present case, positive MHC-1 and C5b9 staining supported the diagnosis of inflammatory myopathy associated with cGVHD, highlighting the need for careful interpretation of the diagnostic findings in such cases.
Previous studies by Shono et al.4 contribute to the understanding of bone marrow GVHD, while Shulman et al.3 focus on the neurologic and cardiac complications associated with GVHD. Lehky et al.6 investigated the prevalence of neuropathy and muscle cramps in patients undergoing HSCT and found that 2.6–8.1% of patients exhibiting cGVHD symptoms may develop neuromuscular complications. Myositis was the most common neuromuscular disorder, followed by neuropathy and, less frequently, myasthenia gravis, and 55% of patients with neuromuscular symptoms experienced muscle cramps, while 65% demonstrated a muscle weakness phenotype.6
Kvinge et al.2 conducted a comprehensive review of the literature on musculoskeletal manifestations of cGVHD and reported that cutaneous (52%) and oropharyngeal (37%) complications were the most common complications of cGVHD, while musculoskeletal complications (0.5%–3%) were among the rarer findings. They also emphasized that muscle pain is a grave and debilitating condition that should be considered in patients exhibiting muscle weakness, joint stiffness, and tissue inflammation.
Rørvik et al.7 provided an overview of different clinical presentations of cGVHD, categorizing fasciitis and joint stiffness as musculoskeletal findings, and myositis and polymyositis as diagnostic indicators; notably, they described cardiac myositis and pericardial involvement as uncommon. Likewise, Tan et al.11 reported a case of polymyositis following stem cell transplantation in a 6-year-old patient with thalassemia. The researchers underline that approximately 40 comparable cases have been acknowledged worldwide, the treatment of which is intricate. Moreover, Allen et al.12 provided clinical, pathological, and molecular examination details of three patients with cGVHD who developed myopathy post-allogeneic HSCT. In each instance, Allen et al.12 noticed perifascicular atrophy, a histological hallmark of dermatomyositis (DM).
There are no reliable benchmarks for the histopathological confirmation of muscle involvement in either aGVHD or cGVHD; however, excluding potential causative factors leading to similar morphological appearances may help determine a confirmed diagnosis. The upregulation of MHC-1 is a significant finding in inflammatory/immune-related myopathies; this is particularly noteworthy for dermatomyopathies, where it is more pronounced in perifascicular regions. However, our patient did not exhibit other typical features of dermatomyopathies, such as perifascicular atrophy, cell reactions with CD20-positive lymphocytes (especially pronounced in the perimysial region), COX deficiency in enzyme stains, and a serological increase in CK. Hence, inflammatory dermatomyopathy was not considered a potential diagnosis.
Other potential differential diagnoses of musculoskeletal manifestations of GVHD include hereditary myopathies (facioscapulohumeral muscular dystrophy [FSHMD], limb girdle muscular dystrophy [LGMD] 2A and 2B, hereditary inclusion body myopathy [HIBM]). In such cases, dystrophic findings from the distribution of cellular reactions in the biopsy specimen, combined with the patient’s personal and family history, may facilitate differentiation. On the other hand, in toxic and drug-induced myopathies, pathological findings attributed to the causative agent are crucial. Notably, steroids, commonly utilized in treating GVHD, can cause steroid-induced myopathy, which is characterized by painless proximal muscle weakness in the bilateral upper and lower extremities. The diagnosis is primarily clinical, with fatty alterations in proximal muscles seen on magnetic resonance imaging. Unlike other myopathies, steroid-induced myopathy is less likely to be detected via EMG5; muscle biopsy often reveals selective atrophy of type II muscle fibers.13 In cases of polymyositis and DM linked to connective tissue diseases, both clinical history and serologic findings play a significant role (Table I).14
| AP: Alkaline phosphatase, cGVHD: Chronic graft versus host disease, CK: Creatine kinease, COX: Cytochrome oxidase, C5b9: complement 5b-9, DM: Dermatomyositis, GI: Gastrointestinal, IM: Immune myopathies, MHC-1: Major Histocompatibility Complex class I, NOS: Nitric oxide synthase. | ||||||||
| Table I. Differential diagnosis in immune dermatomyopathy syndromes.14 | ||||||||
| Features | IM + perimysial pathology | Dermatomyositis + vascular pathology | Regional ischemic immune myopathy | DM: pauci-myopathic | cGVHD |
Myositis + Mi-2 antibody |
Systemic sclerosis | |
| Clinical | Age at onset | Adult > child | Child or adult | Older adult | Adult | Adult | Child or adult | Adult |
| Weakness | Proximal > distal | Proximal > distal | Proximal > distal | Uncommon | Proximal ± distal (50%) | Proximal (98%) | Proximal | |
| Myalgia | Common | Common | Common | None or Mild | Some | |||
| Skin pathology | Raynaud | Rash | Rash (70%) | Palmar: papules & ulcers | Sclerosis | Rash | Scleroderma | |
| Rash | Heliotrope | MxA + Keratinocytes | Dyskeratosis | Raynaud | Raynaud | |||
| Mechanic's hands | Limbs: Extensor surface | Type I IFN | Capillary telangiectasia | |||||
| Psoriasiform | Capillary Δ Tif1γ: Photosensitive | Alopecia | ||||||
| Eczematous | Palm hyperkeratosis | |||||||
| Dyskeratosis | NXP2: Edema | |||||||
| Interstitial lung disease | Common | Uncommon | No | Common | Common | Rare | Some | |
| Neoplasm association | No | Adults | Common; 72%, > 60 yrs. | No | Prior, hematologic | No | No | |
| Systemic, other | Joints | Calcification; GI | GI; mucous membranes | Joints; GI |
Esophagus, small bowel Calcinosis |
|||
| Serum | Myositis-associated antibody |
Jo-1 tRNA synthetase |
TIF1γ NXP-2 |
TIF1γ NXP-2 |
MDA5 | Varied | Mi-2 |
HEp-2 IIF nuclear SMN |
| CK > 1,000 U/L | Some | Few | Most | No | No | Most | Some | |
| Muscle pathology | Inflammation | No | No | Uncommon | Yes (50%) | No | ||
| Focal | Some | Yes (80%) | ||||||
| Location | Perimysium | Perivascular | Perimysial & Endomysial | |||||
| Type | Histiocytic | Lymphocyte (B-cell) | Lymphocyte | |||||
| Perimysium pathology | No | |||||||
| Fragmentation | Common | Occasional | Necrotic regions | Some | Common | |||
| Immune cells | Histiocytic Scattered | Lymphocyte (B-cell) perivascular | Histiocytic |
Histiocytic Scattered |
Histiocytic | Histiocytic | ||
| AP | Some | Uncommon | Yes | Some | Common | |||
| Myofibril pathology | ||||||||
| General | Mild or no | Glycosylation ↓ | ||||||
| Perifascicular | Often | Yes | No | NOS2 | No | Yes | MHC-1 upregulated | |
| COX staining reduced | Never | Most patients | No | |||||
| Necrosis & regeneration | Common | Unusual | ||||||
| LC-3 aggregates | Rare | Common | Common 60% | |||||
| Atrophy | Uncommon | Common | Perifascicular | |||||
| Necrosis | Regional | No | No | |||||
| Capillaries near myofiber | Normal | Reduced | Normal | Reduced | Normal | Thick walls | ||
| C5b-9 | Normal | Large | Large | |||||
| AP stain | Absent | Few | Common | |||||
| Large vessel pathology | No | Artery + vein | Vein | No | No | No | No | |
In conclusion, this case report describes the features of a rare case of musculoskeletal GVHD with DM-like manifestations in a pediatric patient undergoing allogeneic HSCT for AML-M2. Through this unique case, we aim to enhance the understanding of atypical GVHD presentations and highlight the need for a comprehensive diagnostic approach for prompt and effective treatment.
Acknowledgements
We would like to thank Prof. Hans-H. Goebel, Prof. Carolina E. Sewry, Prof. Erdener Özer and Prof. Gülden Diniz for sharing their scientific knowledge with us.
Ethical approval
Informed consent was obtained from the patient’s parents. They also agreed to the publication of this case.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Justiz Vaillant AA, Modi P, Mohammadi O. Graft-Versus-Host Disease. In: StatPearls. Treasure Island (FL): StatPearls Publishing; Oct 10, 2022.
- Kvinge AD, Kvammen T, Miletic H, Bindoff LA, Reikvam H. Musculoskeletal chronic graft versus host disease-a rare complication to allogeneic hematopoietic stem cell transplant: a case-based report and review of the literature. Curr Oncol 2022; 29: 8415-8430. https://doi.org/10.3390/curroncol29110663
- Shulman HM, Cardona DM, Greenson JK, et al. NIH Consensus development project on criteria for clinical trials in chronic graft-versus-host disease: II. The 2014 Pathology Working Group Report. Biol Blood Marrow Transplant 2015; 21: 589-603. https://doi.org/10.1016/j.bbmt.2014.12.031
- Shono Y, Ueha S, Wang Y, et al. Bone marrow graft-versus-host disease: early destruction of hematopoietic niche after MHC-mismatched hematopoietic stem cell transplantation. Blood 2010; 115: 5401-5411. https://doi.org/10.1182/blood-2009-11-253559
- Smith SR, Haig AJ, Couriel DR. Musculoskeletal, neurologic, and cardiopulmonary aspects of physical rehabilitation in patients with chronic graft-versus-host disease. Biol Blood Marrow Transplant 2015; 21: 799-808. https://doi.org/10.1016/j.bbmt.2014.10.019
- Lehky T, Fernandez IP, Krakow EF, et al. Neuropathy and muscle cramps in autologous and allogeneic hematopoietic cell transplantation survivors. Transplant Cell Ther 2022; 28: 608.e1-608.e9. https://doi.org/10.1016/j.jtct.2022.06.009
- Rørvik SD, Abrahamsen IW, Myhre AE, et al. Chronic graft-versus-host disease. Tidsskr Nor Laegeforen 2023; 143: 10.4045/tidsskr.22.0525. https://doi.org/10.4045/tidsskr.22.0525
- Couriel DR, Beguelin GZ, Giralt S, et al. Chronic graft-versus-host disease manifesting as polymyositis: an uncommon presentation. Bone Marrow Transplant 2002; 30: 543-546. https://doi.org/10.1038/sj.bmt.1703711
- New-Tolley J, Smith C, Koszyca B, et al. Inflammatory myopathies after allogeneic stem cell transplantation. Muscle Nerve 2018; 58: 790-795. https://doi.org/10.1002/mus.26341
- Oda K, Nakaseko C, Ozawa S, et al. Fasciitis and myositis: an analysis of muscle-related complications caused by chronic GVHD after allo-SCT. Bone Marrow Transplant 2009; 43: 159-167. https://doi.org/10.1038/bmt.2008.297
- Tan Y, Lin J, Hong X, Lu J, Lu Q. Polymyositis in a child with thalassemia after hematopoietic stem cell transplantation: a case report. Medicine (Baltimore) 2021; 100: e27388. https://doi.org/10.1097/MD.0000000000027388
- Allen JA, Greenberg SA, Amato AA. Dermatomyositis-like muscle pathology in patients with chronic graft-versus-host disease. Muscle Nerve 2009; 40: 643-647. https://doi.org/10.1002/mus.21353
- Stevens AM, Sullivan KM, Nelson JL. Polymyositis as a manifestation of chronic graft-versus-host disease. Rheumatology (Oxford) 2003; 42: 34-39. https://doi.org/10.1093/rheumatology/keg025
- Neuromuscular Disease Center. Immune dermatomyopathies: comparative features. St. Louis, MO, USA: Washington University. Available at: https://neuromuscular.wustl.edu/pathol/jo1.htm#dmvsimp
Copyright and license
Copyright © 2025 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









