
Graphical Abstract
Abstract
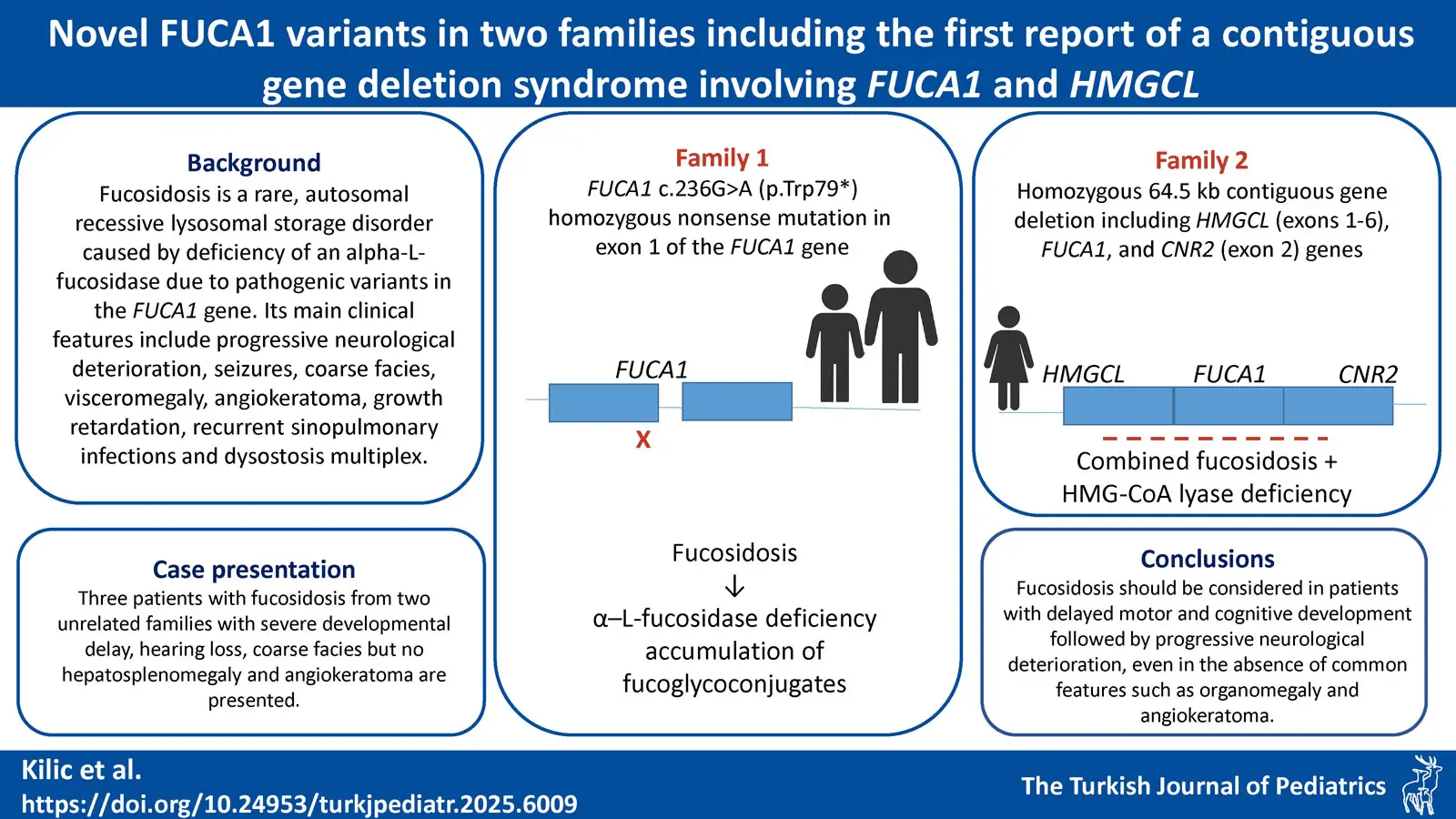
Background: Fucosidosis is a rare, autosomal recessive lysosomal storage disorder caused by deficiency of an alpha-L-fucosidase due to pathogenic variants in the FUCA1 gene, leading to the accumulation of fucoglyco-conjugates in the lysosomes of the liver, brain, skin and other organs. Its main clinical features include progressive neurological deterioration, seizures, coarse facies, visceromegaly, angiokeratoma, growth retardation, recurrent sinopulmonary infections and dysostosis multiplex.
Case Presentation. Three patients with fucosidosis from two unrelated families with severe developmental delay, hearing loss, coarse facies but no hepatosplenomegaly and angiokeratoma are presented. A homozygous, novel nonsense mutation c.236G>A (p.Trp79*) in exon 1 of the FUCA1 gene was identified in one family, and a homozygous novel 64.5 kb deletion, including HMGCL (exons 1-6), FUCA1, and CNR2 (exon 2) genes in the other.
Conclusions: Fucosidosis should be considered in patients with delayed motor and cognitive development followed by progressive neurological deterioration, even in the absence of common features such as organomegaly and angiokeratoma. The pathogenic variants identified in both families were novel and consistent with fucosidosis type 1. To our knowledge, this is the first reported case of fucosidosis accompanied by 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) lyase deficiency resulting from a contiguous gene deletion involving the HMGCL gene at the 1p36.11 locus.
Keywords: fucosidosis, alpha-L-fucosidase, HMG-CoA lyase deficiency, developmental delay, lysosomal storage disorder, 1p36.11 deletion
Introduction
Fucosidosis (OMIM #230000) is a rare, autosomal recessive neurodegenerative lysosomal storage disorder characterized by a deficiency of alpha-L-fucosidase due to pathogenic variants in the FUCA1 gene (MIM *612280) located on chromosome 1p36.11. Alpha-L-fucosidase (EC 3.2.1.51) hydrolyzes fucose resides from glycolipids, glycoproteins and oligosaccharides. Its deficiency leads to the accumulation of oligosaccharides rich in alpha-L-fucose in many organs, particularly in the brain which are excessively excreted in urine.1-8 Clinical features include neurological signs such as progressive psychomotor retardation, seizures, spasticity, and dystonia, along with coarse facies, visceromegaly, angiokeratomas, growth retardation, recurrent respiratory tract infections, and dysostosis multiplex.1-3 Kyphoscoliosis, joint contractures, telangiectasia, hernia, hearing impairment and ophthalmological abnormalities (dilated and tortuous retinal veins and conjunctival vessels, corneal opacities, pigmentary retinopathy, diminished visual acuity) are also reported.1-3 Previously, the disease was classified according two phenotypes based on age of onset and severity. Type 1 (severe form) follows a rapidly progressive neurodegenerative course with symptoms at the age before 1 to 2 years of age and death occurring in the first decade. Whereas type 2 (milder form) follows a slower neurologic deterioration with symptoms at the age of after 1 to 2 years and survive into the second or third decade of life.3-5 It is now considered as a continuous clinical spectrum with variable severity of phenotype.3-5 Its frequency is below 1 in 200,000 live births.6 The highest incidence has been reported in Italians and the Mexican-Indian or Hispanic-American population.3-8 The diagnosis is based on clinical suspicion, supported by appropriate clinical and radiological examinations followed by urinary examination for oligosaccharide excretion and then specific enzyme assay, usually on white blood cells and genetic analysis. Brain magnetic resonance imaging (MRI) findings include bilateral globus pallidus hyperintensities on T1-, marked hypointensity accompanying hyperintense curvilinear streak between their medial and lateral segments on T2-weighted images and diffuse symmetric white matter hyperintensities on T2-weighted images with normal appearance on T1-weighted images, indicative of hypomyelination.9-11 Brain magnetic resonance imaging spectroscopy (MRS) findings include both the peak at the 3.8–3.9 ppm on being most prominent with short TE values (TE 30/35) that is broadening second creatinine peak, suggesting oligosaccharides and glycolipids and a doublet peak at the 1.2 ppm that inverts on TE 135, suggesting fucose peak. Although there is no specific treatment, patients have reported benefiting from bone marrow transplantation.12 Earlier transplant has been reported to be more effective than transplant after full clinical manifestation. Here we present three fucosidosis type 1 patients from two unrelated families. This report includes the first case of fucosidosis accompanied by 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) lyase deficiency due to a contiguous gene deletion, along with two novel pathogenic variants in the FUCA1 gene.
Materials and Methods
In Patients 1 and 2, Sanger sequencing analysis of the FUCA1 gene was performed using genomic DNA extracted from peripheral blood lymphocytes. The description of the variants utilized the human genome version 38 (hg38) and the transcript NM_000147.5 (RefSeq).
For Patient 3, DNA extracted from peripheral blood was used to perform a chromosomal microarray study using Infinium Global Screening Array Cyto (GSA-Cyto) chips on the Illumina iScan platform. Copy number variations (CNVs) were detected and visualized using the NxClinical (v.6.0) analysis software developed by Biodiscovery. The relevant positions were reported based on the Human Genome Build 37 (GRCh37/hg19) reference. The obtained data were evaluated using current databases (PubMed, OMIM, DGV, DECIPHER).
Detected SNVs and CNVs are classified based on the recommendations of ACMG-AMP 2015 and ACMG-ClinGen 2020 guidelines, respectively.13,14
Case presentations
Patients 1 and 2
A six-year-old Turkish male patient of Uzbek origin in Afghanistan was referred to our clinic due to an inability to sit and speak. His complaints began after the age of one; he had one seizure at the age of two, but was not started on medication as it did not recur. He was able to sit at the age of 12 months, but he was never able to stand, walk or speak. The parents were first-degree cousins. At presentation, he was spastic tetraplegic with growth retardation and could speak no meaningful words. He had dystonia triggered by stimuli. His deep tendon reflexes were brisk. He had a depressed nasal bridge and coarse facial features. Skin examination was normal and there was no organomegaly. X-ray revealed a dysostosis multiplex including anterior beaking of vertebrae, lumbar hyperlordosis, widened and scalloped acetabular roof. Brain MRI showed marked hypointensity in both globi pallidi, diffuse symmetric white matter hyperintensities on T2-weighted images, cerebellar folia and cerebral atrophy with enlargement of the lateral ventricles. Electroencephalography showed epileptic activity. Ophthalmologic examination was normal, but the brainstem auditory evoked potentials test was bilaterally pathological. Tandem mass spectrometry, quantitative blood amino acid analysis, biotinidase activity, homocysteine, very long-chain fatty acids, transferrin isoelectric focusing and urine organic acid analysis were all in normal range. Urine glycosaminoglycan analysis was normal, but urine thin-layer chromatography showed increased oligosaccharides, particularly consistent with fucosidosis. The alpha-L-fucosidase enzyme level was measured at 1.9 nmol/hour/mg protein (N: >40), which was significantly low, diagnostic of fucosidosis. He is now 9.5 years old, responds to sound, has eye tracking, but is bedridden and fed with a gavage diet.
The patient’s two-year-old male sibling with coarse facial features, developmental delay, hearing loss and dysostosis multiplex was also diagnosed with the same condition through enzymatic and molecular genetic analysis. His deep tendon reflexes were brisk. He had a depressed nasal bridge and coarse facial features. Skin examination was normal and there was no organomegaly. X-ray revealed a dysostosis multiplex including anterior beaking of vertebrae, lumbar hyperlordosis, widened and scalloped acetabular roof. He was able to sit at the age of 12 months and walk at the age of 1.5. He has eye tracking and can respond to sound. However bilateral mild hearing loss was detected and hearing aids were planned. Ophthalmologic examination and electroencephalography were normal. He is now 5.5 years old and has been hospitalized 3 times due to sinopulmonary infection. He can still walk but cannot run. He can only say ‘mom’ and ‘dad’. He can feed himself but has started to lose weight (Table I).
| a: Patients marked with the same superscript letter are siblings. | ||||
| Table I. Comparison of clinical findings in our patients and literature | ||||
| Clinical Finding |
|
(Family 1) |
(Family 1) |
(Family 2) |
| Intellectual disability |
|
|
|
|
| Neurologic regression |
|
|
|
|
| Coarse face |
|
|
|
|
| Growth retardation |
|
|
|
|
| Recurrent infections |
|
|
|
|
| Dysostosis multiplex |
|
|
|
|
| Angiokeratoma |
|
|
|
|
| Seizure |
|
|
|
|
| Organomegaly |
|
|
|
|
| Hearing loss |
|
|
|
|
| Hernia |
|
|
|
|
| Loss of visual acuity |
|
|
|
|
Patients 1 and 2 were found to have the same homozygous variant in exon 1 of the FUCA1 (NM_000147.5) gene, c.236G>A p.Trp79*, which to our knowledge has not been previously documented in the literature. It is considered as a null variant in a gene where loss of function is recognized as a mechanism of the disease. Additionally, it has an extremely low frequency in gnomAD population databases, further supporting its potential clinical significance. It was classified as “pathogenic” based on recommendations of ACGM 2015 criteria with the evidence codes PVS1, PM2, PP4.13 Due to the autosomal recessive inheritance of the disease, genetic counseling was provided to the family and parental segregation analysis is planned.
Patient 3
A 3-year-old Turkish female patient previously followed up with a diagnosis of HMG-CoA lyase deficiency (OMIM #246450) at another hospital, was referred to us with complaints of macrocephaly, coarse face, hearing loss and severe intellectual disability. There were no specific features in the perinatal history. The parents were first-degree cousins and one of their previous children had died at age 2-year-and 6-month due to HMG-CoA lyase deficiency. The patient achieved head control at 4 months, sat with support at 8 months and was able to stand with support at 12 months, with no meaningful words, however she lost motor and cognitive skills following a febrile infection at 12 months. Physical examination revealed normal growth parameters, coarse facies, macrocephaly, pectus carinatum, absent head control and speech, spasticity and pes cavus in the lower extremities. She used a hearing aid. Ophthalmologic examination was normal (Table I). Tandem mass spectrometry revealed significantly elevated C6DC levels and urine organic acid analysis showed marked excretion of 3-methylglutaric acid, 3-methylglutaconic acid, and 3-hydroxy-3-methylglutaric acid, consistent with the diagnosis of HMG-CoA lyase deficiency. Brain MRI showed thinning of the corpus callosum, signal increase in bilateral frontoparietotemporal subcortical areas, all deep white matter, internal and external capsules on T2-weighted images. Although treatment was initiated early and the patient did not experience an acute attack, she developed severe developmental delay, hearing loss, coarse facial features, and macrocephaly which were unexpected findings. Because PCR amplification of exons 1-6 of the HMGCL gene was absent, chromosomal microarray analysis was performed to investigate the presence of a deletion. Chromosomal microarray analysis revealed the homozygous 64.5 kb deletion arr[GRCh37] 1p36.11 (24137225_24201779)x0 in the patient, which includes exons 1-6 of the HMGCL (NM_000191) gene, the whole FUCA1 (NM_000147) gene, and exon 2 of the CNR2 (NM_001841) gene (Fig. 1A). Loss-of-function mutations in both HMGCL and FUCA1 are classified as pathogenic and are related to HMG-CoA lyase deficiency, and fucosidosis, respectively. Due to its complete overlap with established haploinsufficient / loss of function-sensitive genes that are exclusively associated with recessive conditions, it is classified as pathogenic according to the ACMG (2020) criteria. Segregation analysis was performed on the parents, and both were found to be heterozygous for the same deletion (Fig. 1B and Fig. 1C). Notably, to the best of our knowledge, the literature does not report any cases of simultaneous homozygous deletion of both the HMGCL and FUCA1 genes. Unfortunately, due to the patient’s passing from an infection urine oligosaccharide analysis and alpha-L-fucosidase enzyme level were not evaluated. In this patient, we considered the coexistence of HMG-CoA lyase deficiency and fucosidosis as the primary contributors to the clinical findings.

Written informed consents were obtained from the parents for the collection and publication of the patients’ clinical information.
Discussion
The first case of fucosidosis was described by Durand et al. in 1966.1 To date, fewer than 150 cases and less than 50 different pathogenic variants in the FUCA1 gene have been reported worldwide (the HGMD database professional 2024.3, www.hgmd.cf.ac.uk; accessed 13 November 2024). Genotype-phenotype correlations are not well defined.4-8 A wide range of severity can occur within the same family. However, as a rule, patients who are homozygous for null alleles such as the gross deletions or premature stop codon variants are expected to have loss of enzyme activity and a more severe phenotype. The majority of variants are intragenic small deletions/insertions (INDELs), nonsense and splice site variants, with fewer missense variants, all of which can be identified through sequence analysis. Previously, seven exonic deletions were reported in the FUCA1 gene. However, loss of both of FUCA1 and HMGCL genes has not been reported in the literature to date. The pathogenic variants detected in both families were novel and consistent with fucosidosis type 1 (severe form).
Importantly, the most striking feature of Patient 3 was the co-occurrence of two distinct inborn errors of metabolism—fucosidosis and HMG-CoA lyase deficiency—resulting from a contiguous gene deletion. While the co-existence of two Mendelian disorders in a single patient is not entirely unprecedented, what makes this case unique is the underlying genomic mechanism. Contiguous gene deletion syndromes in inherited metabolic disorders are exceedingly rare. Well-documented examples include glycerol kinase deficiency combined with Duchenne muscular dystrophy, and hypotonia-cystinuria syndrome, as well as various mitochondrial DNA deletions. To our knowledge, this is the first report of a contiguous gene deletion encompassing both FUCA1 and HMGCL, leading to the simultaneous manifestation of two metabolic disorders. This finding underscores the importance of considering large-scale genomic rearrangements in patients with atypical or overlapping metabolic phenotypes, and it expands the spectrum of known contiguous gene deletion syndromes in the field of inborn errors of metabolism.
HMG-CoA lyase deficiency is an autosomal recessive disorder caused by biallelic pathogenic variants in HMGCL. HMG-CoA lyase deficiency typically presents with recurrent episodes of encephalopathy including hypoglycemia, metabolic acidosis, vomiting and a reduced level of consciousness. Most patients present in the first year of life. Neurological problems are common, particularly in neonatal-onset cases. A moderate protein or leucine restriction, low fat diet and carnitine supplements are usually preferred in the management. The genotypes poorly correlate with the clinical phenotype.15 Metabolic investigations in the third patient were consistent with HMG-CoA lyase deficiency.
Fucosidosis should be considered in the differential diagnosis of patients with progressive neurologic deterioration. All three patients had severe developmental delay with neuromotor regression beginning at one year of age. Coarse facies, hearing loss and dysostosis multiplex were present in all of the patients. Hepatosplenomegaly and angiokeratoma were not found in any of the patients. Willems et al. reported visceromegaly and angiokeratoma in 30% and 52% of patients, respectively.3
In Patient 3, HMG-CoA lyase deficiency was diagnosed at the early neonatal period as the first diagnosis by selective screening due to an affected sibling history after which early treatment was started. Although she did not have metabolic attacks, her developmental milestones were significantly delayed. In addition, she had coarse facies, macrocephaly, hearing loss and dysostosis multiplex inconsistent with HMG-CoA lyase deficiency. Genetic studies were performed both to confirm HMG-CoA lyase deficiency and to investigate the possible second genetic disorder. Two inborn errors of metabolism including HMG-CoA lyase deficiency and fucosidosis were identified by a chromosomal microarray analysis. Therefore, comprehensive genetic and metabolic testing should be pursued in cases where clinical findings cannot be explained by a single disorder.
One of the key limitations of this study is the inability to perform enzymatic analysis and urinary oligosaccharide testing for Patient 3, as the patient was deceased. However, the clinical findings including macrocephaly, coarse facial features, and hearing loss strongly support the diagnosis of fucosidosis. Genetic analysis confirmed the diagnosis by identifying a homozygous deletion in the patient, with both parents being heterozygous carriers. The CNR2 gene has not been associated with any Online Mendelian Inheritance in Man (OMIM) phenotype to date. The potential contribution of the CNR2 deletion to a clinical entity with overlapping features cannot be entirely ruled out. However, this hypothesis requires further functional studies and additional experiments. Both families have received counseling regarding prenatal diagnostic options and the importance of genetic testing in future pregnancies.
In conclusion, angiokeratoma and hepatosplenomegaly may not be consistent findings in fucosidosis. When unexpected new findings emerge, the possibility of a second disease should be considered and additional diagnostic tests should be performed. To our knowledge, this is the first reported case in the literature of fucosidosis accompanied by HMG-CoA lyase deficiency resulting from a contiguous gene deletion, with merged phenotypes of both disorders.
Ethical approval
A signed informed consent form was obtained from each participant’s parents for the publication of this report.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Durand P, Borrone C, Della Cella G. A new mucopolysaccharide lipid storage disease? Lancet 1966; 288: 1313-1314. https://doi.org/10.1016/S0140-6736(66)91718-1
- Simon J, Frits AW. Glycosaminoglycans and oligosaccharides disorders: glycosaminoglycans synthesis defects, mucopolysaccharidoses, oligosaccharidoses and sialic acid disorders. In: Saudubray JM, Baumgartner MR, Ángeles García-Cazorla, Walter JH, editors. Inborn Metabolic Diseases Diagnosis and Treatment. 7th ed. Germany: Springer Nature; 2022: 765-783. https://doi.org/10.1007/978-3-662-63123-2_41
- Willems PJ, Gatti R, Darby JK, et al. Fucosidosis revisited: a review of 77 patients. Am J Med Genet 1991; 38: 111-131. https://doi.org/10.1002/ajmg.1320380125
- Stepien KM, Ciara E, Jezela-Stanek A. Fucosidosis-clinical manifestation, long-term outcomes, and genetic profile-review and case series. Genes (Basel) 2020; 11: 1383. https://doi.org/10.3390/genes11111383
- Willems PJ, Seo HC, Coucke P, Tonlorenzi R, O’Brien JS. Spectrum of mutations in fucosidosis. Eur J Hum Genet 1999; 7: 60-67. https://doi.org/10.1038/sj.ejhg.5200272
- Gowda VK, Srinivasan VM, Vegda H, Bhat M. Fucosidosis with pathogenic variant in FUCA1 gene. Indian J Pediatr 2020; 87: 867-868. https://doi.org/10.1007/s12098-020-03246-7
- do Rosario MC, Purushothama G, Narayanan DL, Siddiqui S, Girisha KM, Shukla A. Extended analysis of exome sequencing data reveals a novel homozygous deletion of exons 3 and 4 in FUCA1 gene causing fucosidosis in an Indian family. Clin Dysmorphol 2023; 32: 112-115. https://doi.org/10.1097/MCD.0000000000000452
- Tiberio G, Filocamo M, Gatti R, Durand P. Mutations in fucosidosis gene: a review. Acta Genet Med Gemellol (Roma) 1995; 44: 223-232. https://doi.org/10.1017/s0001566000001641
- Galluzzi P, Rufa A, Balestri P, Cerase A, Federico A. MR brain imaging of fucosidosis type I. AJNR Am J Neuroradiol 2001; 22: 777-780.
- Autti T, Joensuu R, Aberg L. Decreased T2 signal in the thalami may be a sign of lysosomal storage disease. Neuroradiology 2007; 49: 571-578. https://doi.org/10.1007/s00234-007-0220-6
- Malatt C, Koning JL, Naheedy J. Skeletal and brain abnormalities in fucosidosis, a rare lysosomal storage disorder. J Radiol Case Rep 2015; 9: 30-38. https://doi.org/10.3941/jrcr.v9i5.2149
- Miano M, Lanino E, Gatti R, et al. Four year follow-up of a case of fucosidosis treated with unrelated donor bone marrow transplantation. Bone Marrow Transplant 2001; 27: 747-751. https://doi.org/10.1038/sj.bmt.1702994
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405-424. https://doi.org/10.1038/gim.2015.30
- Riggs ER, Andersen EF, Cherry AM, et al. Correction: Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med 2021; 23: 2230. https://doi.org/10.1038/s41436-021-01150-9
- Morris AAM. Disorders of ketogenesis and ketolysis. In: Saudubray JM, Baumgartner MR, Ángeles García-Cazorla, Walter JH, editors. Inborn Metabolic Diseases Diagnosis and Treatment. 7th ed. Germany: Springer Nature; 2022: 303-313. https://doi.org/10.1007/978-3-662-63123-2_13
Copyright and license
Copyright © 2025 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









