
Graphical Abstract
Abstract
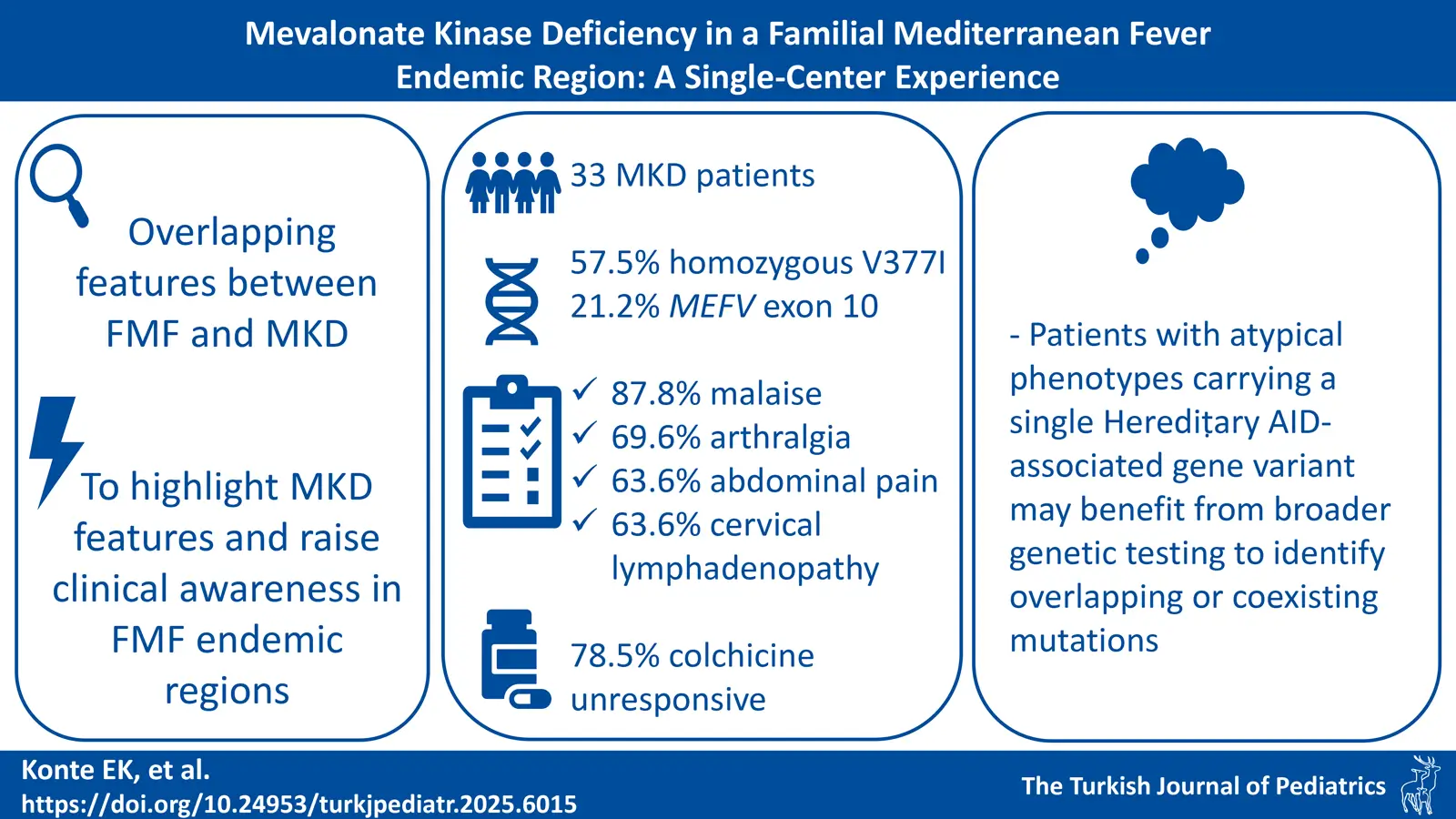
Background. We aimed to document childhood onset mevalonate kinase deficiency (MKD) and to explore treatment responses and diagnostic challenges in regions endemic to familial Mediterranean fever (FMF).
Methods. This retrospective study included patients under 18 years of age, diagnosed with MKD and followed for at least six months at the pediatric rheumatology department of Istanbul University - Cerrahpaşa Medical Faculty between 2016 and 2024.
Results. Of 33 patients, 51.5% were female, with a median age of symptom onset at 6 (2-17.3) months. Eight patients had a history of tonsillectomy, and seven exhibited an underlying exon 10 Mediterranean FeVer (MEFV) gene mutation. The mean diagnostic delay was 67.6 months, which was longer for those with exon 10 mutations (95.0 months) and those with a history of tonsillectomy (99.5 months). The median duration of attacks was 5 (4-7) days. The median frequency of attacks was 12 (10-24) per year. The most prevalent clinical findings observed during these attacks included malaise (87.8%), arthralgia (69.6%), abdominal pain (63.6%), cervical lymphadenopathy (63.6%), diarrhea (54.5%), and maculopapular rash (51.5%). A total of 30 patients (90.9%) identified pre-attack triggers. Among the patients evaluated, 19 (57.5%) were homozygous for V377I, and 7 (21.2%) had V377I biallelic heterozygous mutation in MVK gene. Cytopenia was observed in 18 patients (54.5%) during episodes, including anemia (39.3%), lymphopenia (24.2%), leukopenia (12.1%), and neutropenia (9%).
Conclusions. Patients presenting with periodic fever suggestive of FMF who exhibit atypical features should be evaluated for MKD. Further genetic testing should be performed when atypical clinical findings are present, even in those carrying pathogenic variants in exon 10 of the MEFV gene.
Keywords: hereditary autoinflammatory diseases, familial Mediterranean fever, mevalonate kinase deficiency, tonsillectomy, periodic fever
Introduction
Recurrent fevers are characterized by inflammatory episodes, interspersed with overall well-being intervals.1 These fevers can arise from various etiologies, including infectious, malignant, autoimmune, inflammatory, and genetic causes, which complicates diagnosis. Following the identification of the MEditerranean FeVer (MEFV) gene in 1997, the concept of autoinflammation was introduced to highlight a group of diseases characterized by abnormal activation of the innate immune system occurring independently of pathogens and without the presence of circulating autoantibodies or self-reactive cells.2,3 Monogenic defects in innate immunity have shown that inflammasome activity and interleukin-1 (IL-1) release are compromised in rare conditions known as hereditary autoinflammatory diseases (HAIDs). They are characterized by recurrent inflammation affecting the skin, joints, gastrointestinal tract, central nervous system, and other tissues, with interleukin (IL)-1β as the primary mediator of inflammation.4 The understanding of HAIDs continues to expand with advancements in sequencing technology, and many recent developments have blurred the lines between autoimmunity, immunodeficiency, and autoinflammation.2
Familial Mediterranean fever (FMF) and mevalonate kinase deficiency (MKD) are the two most common HAIDs, sharing overlapping features such as recurrent fever, abdominal pain, arthralgia, and arthritis.3,5 FMF is particularly prevalent in high-risk populations—including Armenians, Sephardic Jews, Turks, and Arabs— where MEFV mutation carrier frequencies range from 1 in 5 to 1 in 7, defining these regions as endemic.4,6-8 In contrast, MKD predominantly affects individuals of Western European descent and results from biallelic mutations in the mevalonate kinase (MVK) gene.9,10 While both diseases share clinical features, MKD is typically characterized by febrile episodes lasting 3 to 7 days, often triggered in infancy—particularly following vaccinations—accompanied by a rash, diarrhea, mucosal ulcers, and cervical lymphadenopathy.5,10,11 Notably, bilateral lymphadenitis may lead to misdiagnosis as periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) syndrome, particularly in patients unresponsive to tonsillectomy.12-14
Despite the development of various classification criteria for HAIDs, diagnostic delay remains common in patients with recurrent inflammatory episodes, increasing the risk of complications such as AA amyloidosis.15 Colchicine is a highly effective therapy for FMF, significantly reducing attack frequency and preventing amyloidosis, even in cases with partial clinical response. Compared to FMF, colchicine has been found to be ineffective in MKD. The prevalence of AA amyloidosis is roughly estimated to be about 6% of all MKD cases.16 The evidence-based therapy for MKD is IL-1 blockade with canakinumab, which is the treatment of choice for patients experiencing frequent disease flares.17
In our study, we aimed to document the clinical characteristics of our patients with childhood-onset MKD, evaluate the treatment response, and investigate the reasons for diagnostic challenges in FMF-endemic regions to raise awareness among clinicians.
Materials and Methods
Study population
This study included patients under the age of 18 years who were diagnosed with MKD and were followed up for at least six months at the pediatric rheumatology department of Istanbul University - Cerrahpaşa Medical Faculty between 2016 and 2024. Among the patients enrolled in the study, 29 individuals met the diagnostic criteria for MKD according to the Eurofever/PRINTO classification for hereditary recurrent fevers.1 These criteria include the presence of either homozygous or biallelic heterozygous mutation in the MVK gene, accompanied by at least one of the following clinical manifestations: gastrointestinal symptoms, cervical lymphadenitis, or aphthous stomatitis.
Additionally, four patients exhibited a heterozygous mutation in the MVK gene and fulfilled the diagnostic criteria for mevalonate kinase deficiency as outlined in the Eurofever/PRINTO clinical classification for PFAPA and hereditary recurrent fevers.1 The criteria for this diagnosis include the presence of at least three of the following six features: age at onset less than one-year, gastrointestinal symptoms, painful lymphadenopathy, aphthous stomatitis, specific triggering factors, and maculopapular rash.
Data collection
Demographic data, including age at onset, age at diagnosis, gender, and family history, were systematically recorded. Clinical manifestations were assessed, focusing on the type and duration of febrile episodes and the frequency of attacks per year. Laboratory evaluations were conducted before and after diagnosis, as well as during the most recent follow-up visit. Laboratory tests included the assessment of inflammatory markers, such as complete blood cell count, C-reactive protein (CRP) level, and erythrocyte sedimentation rate (ESR), along with mutation analysis. Next-generation sequencing analysis method was performed for genetic analysis. Based on the results of the genetic testing, MVK genotypes were documented in order of frequency. Previous diagnoses and treatment regimens were also noted, and the number of attacks following the initiation of treatment was recorded. Our center did not measure immunoglobulin D or urine mevalonate during episodes.
The standard colchicine dose was adjusted according to patients’ ages, following EULAR recommendations for the management of FMF: ≤ 0.5 mg/day for children under 5 years, 0.5–1.0 mg/day for children aged 5–10 years, and 1.0–1.5 mg/day for those over 10 years of age, including adults. We assessed the response to colchicine over a follow-up period of at least 6 months, in line with EULAR recommendations.18
Anakinra was initially administered subcutaneously at a dosage of 2 mg/kg for patients weighing ≤50 kg or 100 mg for those over 50 kg, once daily, according to our clinic’s drug label. For patients in clinical remission, dosing was extended after one month, with a transition to canakinumab after one to three months. Canakinumab was prescribed subcutaneously at a dose of 2-4 mg/kg for patients weighing ≤40 kg or 150 mg for those over 40 kg, every eight weeks. For patients in clinical remission, the dosing interval was further extended after six months.
Statistical analysis
Statistical analyses were performed using IBM SPSS Statistics for Windows, Version 28, and figures were generated using GraphPad Prism software. Categorical variables were presented as frequencies and percentages. Continuous variables were expressed as mean ± standard deviation (SD) if normally distributed, and as median with interquartile range (IQR, 25th–75th percentiles) if not. The distribution of continuous variables was assessed using the Kolmogorov–Smirnov test. Comparisons between groups (patients with and without exon 10 MEFV mutations) were conducted using the chi-square test or Fisher’s exact test for categorical variables, as appropriate. For continuous variables, the Mann–Whitney U test was used. A p-value < 0.05 was considered statistically significant.
Ethical approval
The study was approved by the Istanbul University-Cerrahpasa Ethics Committee (E-83045809- 604.01-910551).
Results
Demographic characteristics and medical history of patients
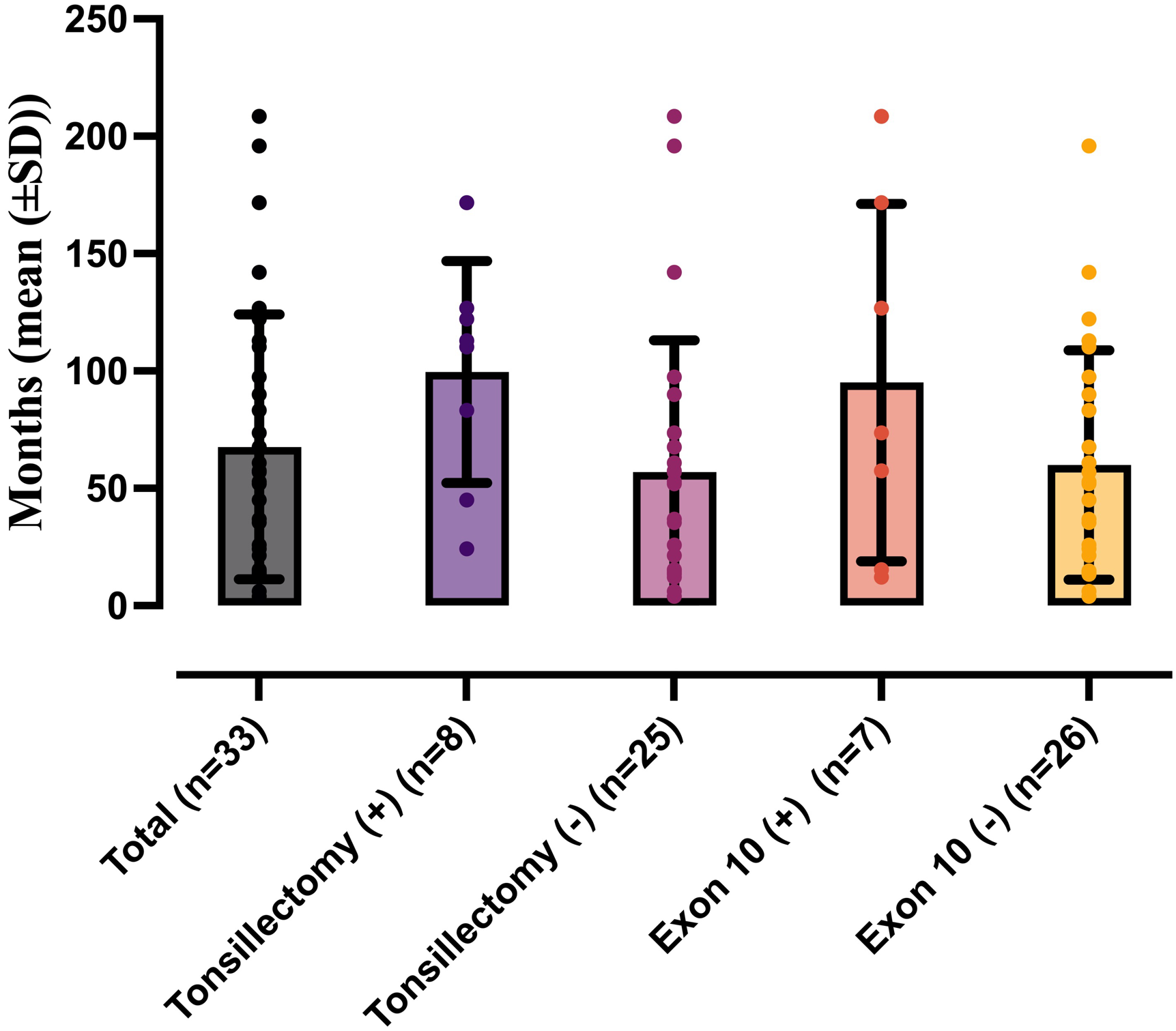
Of the 33 patients, 17 (51.5%) were female, and 16 (48.5%) were male. The median age at symptom onset was 6 (2-17.3) months. The mean age at diagnosis, follow-up period, and age at the last visit were 86.7±57.6, 47.5±46.5, and 134.3±67.2 months, respectively (Table I).
| Data presented as n (%), mean ± standard deviation, or median (Q1-Q3); MEFV: Mediterranean fever; MVK: mevalonate kinase. | |
| Table I. Demographic characteristics of patients with mevalonate kinase deficiency (N=33). | |
| Gender | |
| Female |
|
| Male |
|
| Age at last visit (months) |
|
| Age at symptom onset (months) |
|
| Age at diagnosis (months) |
|
| Follow up duration (month) |
|
| Diagnostic delay (months) |
|
| MEFV (+) (n=7) |
|
| MEFV (-) (n=26) |
|
| Tonsillectomy (+) (n=8) |
|
| Tonsillectomy (-) (n=25) |
|
| MVK genotype | |
| V377I/V377I |
|
| V377I/another MVK |
|
| Another MVK |
|
| With exon 10 MEFV mutation |
|
Among the patients, 12 (36.3%) reported a family history of periodic autoinflammatory diseases: two with MKD alone, two with both MKD and FMF; and eight with FMF alone. Eight patients (24.2%) underwent tonsillectomy for PFAPA syndrome prior to the diagnosis of MKD.
The mean diagnostic delay was 67.6±56.4 months. This delay was prolonged to 95.0±76.2 months in patients with MEFV exon 10 mutation, compared to 58.4 ±48.4 months in those without such mutations. The median diagnostic delay for patients with a history of tonsillectomy was 99.5±47.2 months, while it was 57±56 months for those without (Table I, Fig. 1).
Clinical features
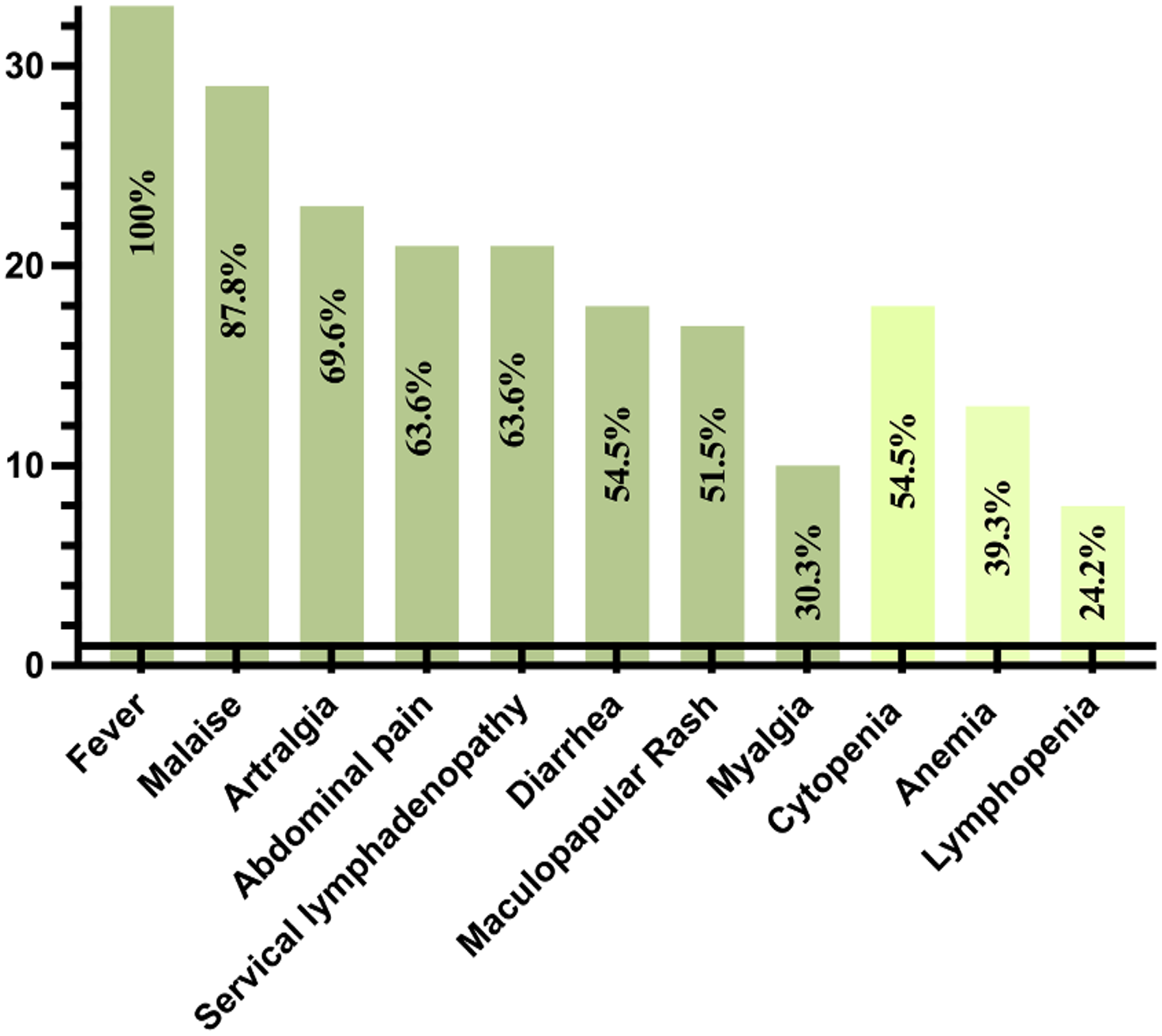
All patients reported fever during the episodes (attacks). The most prevalent clinical findings observed during these attacks included malaise in 29 patients (87.8%), arthralgia in 23 (69.6%), abdominal pain in 21 (63.6%), and cervical lymphadenopathy in 21 (63.6%). Additionally, diarrhea was reported in 18 patients (54.5%), and maculopapular rash was observed in 17 patients (51.5%) (Fig. 2, Table II). Less frequently noted symptoms included myalgia (n=10; 30.3%), vomiting (n=6; 18.1%), oral aphthae (n=4; 12.1%), headache (n=4; 12.1%), febrile seizures (n=3; 9%), arthritis (n=2; 6%), chest pain (n=2; 6%), and hepatosplenomegaly (n=2; 6%). One patient was diagnosed with renal failure resulting from renal AA amyloidosis.
|
Data presented as n (%), or median (Q1-Q3). CRP: C-reactive protein; ESR: erythrocyte sedimentation rate; MEFV: Mediterranean fever; MVK: mevalonate kinase; WBC: white blood cell. |
||||
| Table II. Comparison of clinical and laboratory features between patients with and without MEFV exon 10 mutations. | ||||
|
|
|
|
|
|
| Clinical symptoms | ||||
| Fever |
|
|
|
|
| Malaise |
|
|
|
|
| Artralgia |
|
|
|
|
| Abdominal pain |
|
|
|
|
| Servical lymphadenopathy |
|
|
|
|
| Diarrea |
|
|
|
|
| Maculopapular rash |
|
|
|
|
| Myalgia |
|
|
|
|
| Cytopenia |
|
|
|
|
| Anemia |
|
|
|
|
| Lymphopenia |
|
|
|
|
| Hemoglobulin (g/dL) |
|
|
|
|
| WBC (106/L) |
|
|
|
|
| Platelet count (106/L) |
|
|
|
|
| ESR (mm/h) |
|
|
|
|
| CRP (mg/L) |
|
|
|
|
| Colchicine response |
|
|
|
|
| Attack duration (days) |
|
|
|
|
| Attack number per year, before diagnosis |
|
|
|
|
| Attack number per year, at last visit |
|
|
|
|
When comparing the clinical manifestations between MEFV exon 10 mutation-positive and -negative subgroups, no statistically significant differences were observed in the frequency of fever, malaise, arthralgia, abdominal pain, cervical lymphadenopathy, diarrhea, or myalgia (p > 0.05). A maculopapular rash appeared less frequently in the MEFV exon 10 mutation-positive group (14.2% vs. 61.5%), but the difference did not reach statistical significance (p = 0.07; Table II).
A total of 30 patients (90.9%) identified a pre-attack trigger, with the majority reporting an infection as a trigger (n=30; 90.9%). Other identified triggers included vaccination (n=22; 66.6%) and stress (n=2; 6%). The median duration of attacks was noted to be 5 (4-7) days. Before treatment, the median frequency of attack was 12 (10-24) per year (Table II).
Laboratory parameters and genetic testing
Among the patients evaluated, their MVK gene analysis results were as follows:
- 19 (57.5%) were homozygous for the V377I mutation.
- 7 (21.2%) had V377I in compound heterozygosity with E93Fs (n=2), I268T (n=2), G18R (n=1), G144V (n=1), or G202R (n=1).
- 3 (9%) were homozygous for other MVK gene mutations, including I268V (n=1), R388X (n=1), and exon 3 deletion (n=1).
- 4 (12.1%) displayed heterozygous MVK mutations, including V377I (n=2), S52N (n=1), and D170D (n=1).
- 7 (21.2%) patients had heterozygous mutations in the MEFV gene, including V726A (n = 4), M694V (n = 1), M694I (n = 1), and M680I (n = 1).
Cytopenia was observed in 18 patients (54.5%) during attacks. Anemia was particularly prevalent, affecting 13 patients (39.3%), while leukopenia was noted in four patients (12.1%), neutropenia in three patients (9%), and lymphopenia in eight patients (24.2%). Patients with cytopenia exhibited normal blood parameters between episodes, and there were no indications of recurrent or refractory infections suggestive of immunodeficiency.
The median white blood cell (WBC) count at diagnosis was 8550/mm³ (6425–11363), and the median platelet count at diagnosis was 315,000/ mm³ (255,000–433,750). The median hemoglobin (Hb) value was 11.5 (10.8-12.4) g/dL. Median C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) values were 12.4 (1-62.2) mg/L and 12.5 (5-41.7) mm/h at diagnosis (Table II).
Hematological findings, including cytopenia, anemia, and lymphopenia, were similarly distributed between groups stratified by MEFV exon 10 mutation status, with no statistically significant differences observed (p > 0.05). Additionally, laboratory parameters such as Hb level, WBC count, platelet count, ESR, and CRP were comparable between the groups (Table II).
Treatment response
Colchicine was initiated as the first-line treatment in 28 patients (84.8%) suspected of having an autoinflammatory disease prior to the confirmation of MKD. Of these patients, 22 (78.5%) were colchicine unresponsive, and six patients (21.5%) were colchicine responsive. The colchicine response rate was 0% among patients with exon 10 MEFV mutations, compared to 28.5% in those without these mutation. Although the response was lower in the exon 10-positive group, the difference was not statistically significant (p = 0.28; Table II).
After the diagnosis, three patients continued colchicine therapy, 12 were started on anakinra, and 18 patients commenced treatment with canakinumab. Of the 12 patients initially receiving anakinra, 11 switched to canakinumab due to its ease of use, while one patient discontinued treatment because of irregular follow-up. During the last visit, 28 patients were on canakinumab, and four were on colchicine (Fig. 3). Among those receiving colchicine therapy, three had homozygous V377I mutations, while one had a heterozygous S52N mutation.
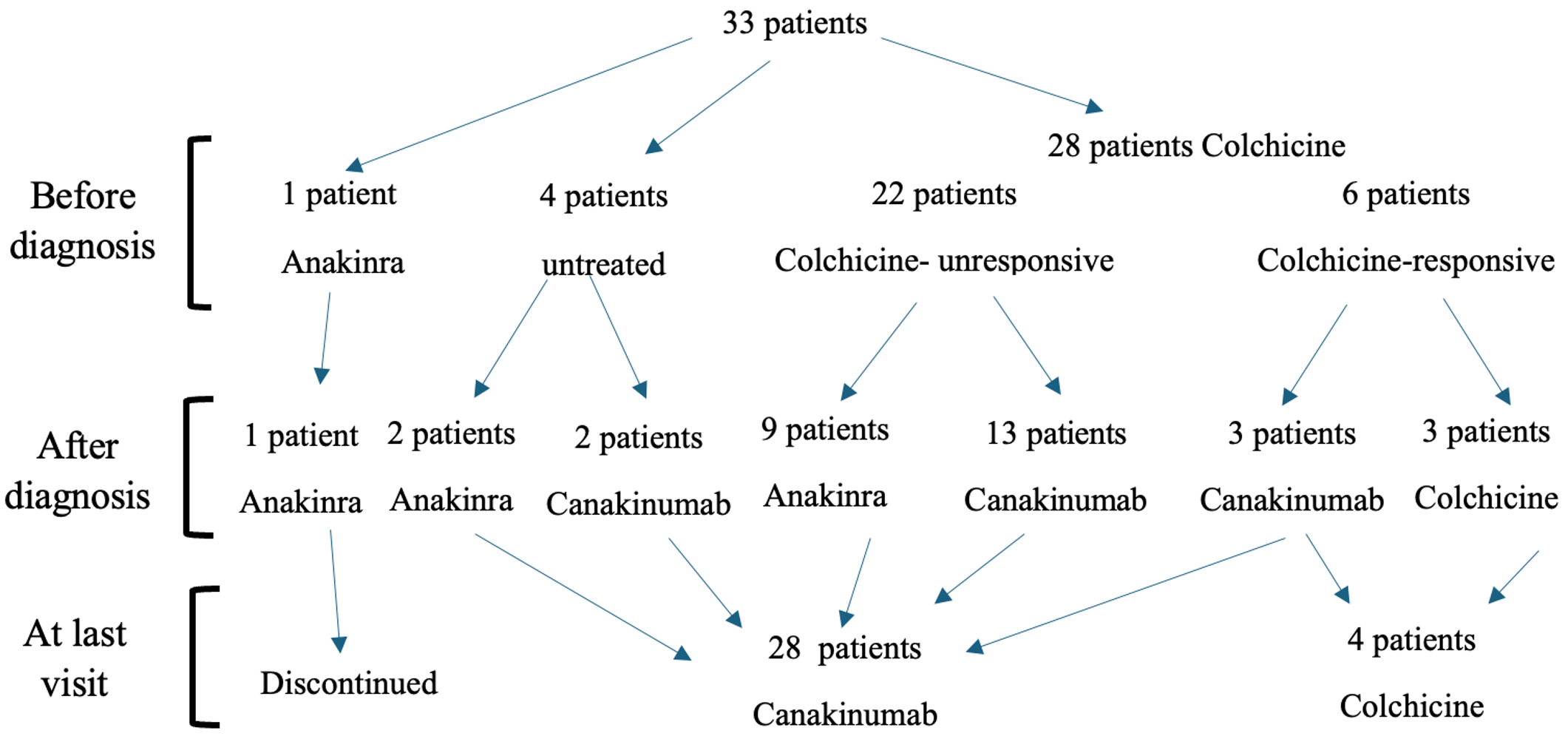
b) After diagnosis: Treatment strategies were adjusted or initiated following molecular diagnosis.
c) At last visit: Therapeutic status at most recent follow-up.
At the final visit, the median number of attacks per year was 1 (0-2). There was no statistically significant difference between the two groups based on the MEFV exon 10 mutation status (p>0.05; Table II). All four patients under colchicine were attack-free at the last visit (one patient had subclinical inflammation). The 28 patients treated with canakinumab received treatment every 2-3 months, and the median number of attacks per year was 1 (0-3).
Discussion
In this retrospective cohort study, we evaluated the clinical features of 33 MKD patients who were followed up in a single center. To our knowledge, this study represents the largest cohort of MKD patients reported in single-center studies so far. Notably, we documented the presence of the MEFV exon 10 variant in seven patients and a history of tonsillectomy due to a diagnosis of PFAPA syndrome in another 8 patients, both of which may contribute to delays in the diagnosis of MKD. Through this research, we aim to enhance awareness regarding the diagnosis of autoinflammatory diseases other than FMF in endemic populations.
Recent advancements in understanding the pathophysiology and genetics of AIDs are facilitating the identification of novel phenotypes. FMF and MKD represent the two most prevalent examples of hereditary autoinflammatory diseases characterized by autosomal recessive inheritance. Specifically, FMF is the most common hereditary autoinflammatory disease among Mediterranean populations, which makes diagnosing rare hereditary autoinflammatory diseases in these groups more challenging.8,12 While MKD is recognized globally, it is more prevalent among individuals of Northern European descent. Mutations in the MVK gene cause MKD by resulting in uncontrolled activation of the pyrin. This triggers caspase-1, resulting in the release of IL-1β, a potent inducer of fever and inflammation.19 Several case reports in the literature describe patients with overlapping features of MKD and FMF.5,20,21 These studies involve patients with a wide range of clinical presentations. In the report by Çakan et al.5 both patients exhibited symptom onset before the age of one and were unresponsive to colchicine therapy. Interestingly, a study by Moussa et al.20 evaluated five siblings carrying mutations associated with both MKD and FMF, and found that only two were symptomatic, while the remaining three were asymptomatic.
PFAPA syndrome is recognised as the most prevalent autoinflammatory disease among children. Although the attacks are frequently manifested by tonsillitis and pharyngitis accompanied by fever, aphthous stomatitis, cervical lymphadenopathy, headache, rash, arthralgia, and abdominal pain may also be observed. A study by Gozen et al.13 reported a response rate to tonsillectomy of 83.2% in patients diagnosed with PFAPA syndrome. In our cohort, eight patients exhibited resistance to tonsillectomy and experienced a longer diagnostic delay for MKD. Therefore, the diagnosis of MKD should be considered in cases of recurrent fevers with atypical presentations who are not responsive to tonsillectomy.14
Our study largely confirms the clinical characteristics of MKD patients described in previous reports. Similarly to studies in the literature, the gender distribution was equal, and the onset age of symptoms was in the first 6 months.22-24 In the study by Hilst et al.22 approximately 50% of 103 MKD patients reported 7 to 12 annual attacks. Ter Haar et al.23 noted a median frequency of 12 attacks per year, while our study recorded a mean of 15.3 attacks per year. The most common genetic mutation was V377I, similar to the literature, and 78% of the patients carried this mutation.22-24 Diagnostic delay begins to decrease as awareness of the disease increases. In the study conducted by Hilst et al.22 the mean diagnostic delay was reported as 13.9 years, with 13 patients (12.6%) being misdiagnosed with FMF. Meanwhile, Ter Haar et al.23 found that the median diagnostic delay was 6 years, which closely aligns with our cohort’s findings.
The most prevalent clinical findings observed during these attacks included malaise (87.8%), arthralgia (69.6%), abdominal pain (63.6%), cervical lymphadenopathy (63.6%), diarrhea (54.5%), and maculopapular rash (51.5%), in accordance with previous reports.22-24 However, in contrast to other studies, oral aphthae, which is also included in the diagnostic criteria for MKD, was found less frequently in our patients. There were no chronic patients except for one with AA amyloidosis at diagnosis, one with an episode of macrophage activation syndrome, and three with epilepsy. In the last published MKD multicenter cohort of 114 patients, one had recurrent macrophage activation syndrome, and five had AA amyloidosis.23
In our study, 22 of 28 patients who were initiated on first-line colchicine treatment were unresponsive at 78.5%. However, at the last follow-up visit, four patients remained free of attacks while on colchicine. In the study by Ter Haar et al.23 colchicine was initiated in 21 patients; one responded, and 13 were unresponsive. Partial response to colchicine was 15.9% in the study by Hilst et al.22 Zhang found a complete non-response of 80% in their literature review.11 Although there are case-based colchicine-responsive cases in the literature25, they have not yet been studied, and studies show that there was no response in the early period. Canakinumab, a fully human anti-IL-1β monoclonal antibody, has been shown to control inflammation and prevent flares effectively in patients with MKD.26-28 In the study by Jeyaratnam et al.19 a median flare rate of 0 was reported, with 83% of patients experiencing either 0 or 1 flare during the study period, compared to a median flare rate of 12 prior to enrollment.
The main limitation of this study is its retrospective nature, consisting of patients with a median disease duration of 47.5±46.5 months. Clinical findings and other data were obtained from the patients’ medical records. The number of patients was insufficient to yield statistically significant results due to the rarity of the disease and the fact that data were collected from a single center. Urine mevalonate, serum amyloid A, and immunoglobulin D levels could not be measured during the attack.
Conclusion
In FMF-endemic regions, FMF remains the most frequently diagnosed autoinflammatory disease in patients with periodic fever. Nevertheless, despite broader awareness and the increasing use of comprehensive genetic panels, FMF is often presumed as the initial diagnosis—even when MEFV mutation status is inconclusive.
Clinicians should be highly suspicious of MKD in colchicine-resistant patients, particularly those with heterozygous MEFV mutations or atypical clinical features. In these cases, further molecular investigations beyond MEFV should be considered. Furthermore, patients with atypical phenotypes who carry a single HAID-associated gene variant may benefit from broader genetic evaluation to identify potential overlapping or coexisting mutations. A more comprehensive and gene-inclusive diagnostic approach may enhance early diagnosis and facilitate the development of targeted treatment strategies.
Ethical approval
The study was approved by İstanbul University-Cerrahpasa Institutional Review Board (date: 09.02.2024, number: 910551).
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Gattorno M, Hofer M, Federici S, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis 2019; 78: 1025-1032. https://doi.org/10.1136/annrheumdis-2019-215048
- Rigante D. New mosaic tiles in childhood hereditary autoinflammatory disorders. Immunol Lett 2018; 193: 67-76. https://doi.org/10.1016/j.imlet.2017.11.013
- Yildiz M, Haslak F, Adrovic A, Barut K, Kasapcopur O. Autoinflammatory diseases in childhood. Balkan Med J 2020; 37: 236-246. https://doi.org/10.4274/balkanmedj.galenos.2020.2020.4.82
- Sangiorgi E, Rigante D. The clinical chameleon of autoinflammatory diseases in children. Cells 2022; 11: 2231. https://doi.org/10.3390/cells11142231
- Çakan M, Aktay-Ayaz N, Keskindemirci G, Karadağ ŞG. Two cases of periodic fever syndrome with coexistent mevalonate kinase and Mediterranean fever gene mutations. Turk J Pediatr 2017; 59: 467-470. https://doi.org/10.24953/turkjped.2017.04.015
- Kisla Ekinci RM, Kilic Konte E, Akay N, Gul U. Familial Mediterranean fever in childhood. Turk Arch Pediatr 2024; 59: 527-534. https://doi.org/10.5152/TurkArchPediatr.2024.24188
- Adrovic A, Yıldız M, Kanber M, et al. Performance of recently proposed periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA) syndrome criteria in a region endemic for familial Mediterranean fever. Rheumatol Int 2020; 40: 91-96. https://doi.org/10.1007/s00296-019-04362-0
- Karacan I, Ugurlu S, Tolun A, Tahir Turanli E, Ozdogan H. Other autoinflammatory disease genes in an FMF-prevalent population: a homozygous MVK mutation and a novel heterozygous TNFRSF1A mutation in two different Turkish families with clinical FMF. Clin Exp Rheumatol 2017; 35 (Suppl. 108): 108: 75-81.
- Rigante D, Frediani B, Cantarini L. A comprehensive overview of the hereditary periodic fever syndromes. Clin Rev Allergy Immunol 2018; 54: 446-453. https://doi.org/10.1007/s12016-016-8537-8
- van der Hilst JC, Frenkel J. Hyperimmunoglobulin D syndrome in childhood. Curr Rheumatol Rep 2010; 12: 101-107. https://doi.org/10.1007/s11926-010-0086-1
- Zhang S. Natural history of mevalonate kinase deficiency: a literature review. Pediatr Rheumatol Online J 2016; 14: 30. https://doi.org/10.1186/s12969-016-0091-7
- Konte EK, Haslak F, Yildiz M, et al. Gray zone in the spectrum of autoinflammatory diseases: familial Mediterranean fever accompanying periodic fever, aphthous stomatitis, pharyngitis, and adenitis syndrome: single-center experience. Eur J Pediatr 2023; 182: 5473-5482. https://doi.org/10.1007/s00431-023-05209-4
- Gozen ED, Yildiz M, Kara S, et al. Long-term efficacy of tonsillectomy/adenotonsillectomy in patients with periodic fever aphthous stomatitis pharyngitis adenitis syndrome with special emphasis on co-existence of familial Mediterranean fever. Rheumatol Int 2023; 43: 137-145. https://doi.org/10.1007/s00296-022-05210-4
- Aktaş B, Gümüş D, Tunalı A, Kunter Z, Adrovic A. Mevalonate kinase deficiency/hyperimmunoglobulin D syndrome (MVK/HIDS) in a differential diagnosis of periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA) syndrome and Familial Mediterranean Fever (FMF): a case report. Turk Arch Pediatr 2022; 57: 365-367. https://doi.org/10.5152/TurkArchPediatr.2022.21321
- Zhang C, Peng J, Liu Z, Zhou Q. Kidney involvement in autoinflammatory diseases. Kidney Dis (Basel) 2023; 9: 157-172. https://doi.org/10.1159/000529917
- Rodrigues F, Philit JB, Giurgea I, et al. AA amyloidosis revealing mevalonate kinase deficiency: a report of 20 cases including two new French cases and a comprehensive review of literature. Semin Arthritis Rheum 2020; 50: 1370-1373. https://doi.org/10.1016/j.semarthrit.2020.03.005
- Jeyaratnam J, Frenkel J. Management of mevalonate kinase deficiency: a pediatric perspective. Front Immunol 2020; 11: 1150. https://doi.org/10.3389/fimmu.2020.01150
- Ozen S, Demirkaya E, Erer B, et al. EULAR recommendations for the management of familial Mediterranean fever. Ann Rheum Dis 2016; 75: 644-651. https://doi.org/10.1136/annrheumdis-2015-208690
- Jeyaratnam J, Simon A, Calvo I, et al. Long-term efficacy and safety of canakinumab in patients with mevalonate kinase deficiency: results from the randomised Phase 3 CLUSTER trial. Rheumatology (Oxford) 2022; 61: 2088-2094. https://doi.org/10.1093/rheumatology/keab696
- Moussa T, Aladbe B, Taha RZ, Remmers EF, El-Shanti H, Fathalla BM. Overlap of familial Mediterranean fever and hyper-IgD syndrome in an Arabic kindred. J Clin Immunol 2015; 35: 249-253. https://doi.org/10.1007/s10875-015-0140-x
- Kousa A, Ahmed R, Abu Bakr MB, Aldosh AN, Khalil B. Complex MEFV and MVK variations in a Syrian child: implications for clinical phenotypes and treatment response-a case report. J Investig Med High Impact Case Rep 2024; 12: 23247096241291929. https://doi.org/10.1177/23247096241291929
- van der Hilst JC, Bodar EJ, Barron KS, et al. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine (Baltimore) 2008; 87: 301-310. https://doi.org/10.1097/MD.0b013e318190cfb7
- Ter Haar NM, Jeyaratnam J, Lachmann HJ, et al. The phenotype and genotype of mevalonate kinase deficiency: a series of 114 cases from the eurofever registry. Arthritis Rheumatol 2016; 68: 2795-2805. https://doi.org/10.1002/art.39763
- Bader-Meunier B, Florkin B, Sibilia J, et al. Mevalonate kinase deficiency: a survey of 50 patients. Pediatrics 2011; 128: e152-e159. https://doi.org/10.1542/peds.2010-3639
- Koç Yekedüz M, Doğulu N, Öncül Ü, Köse E, Ceylaner S, Eminoğlu FT. An atypical presentation of mevalonate kinase deficiency in response to colchicine treatment. Mol Syndromol 2022; 13: 146-151. https://doi.org/10.1159/000518825
- Kilic Konte E, Akay N, Gul U, et al. Long-term safety profile and secondary effectiveness of canakinumab in pediatric rheumatic diseases: a single-center experience. Expert Opin Drug Saf 2025; 24: 915-923. https://doi.org/10.1080/14740338.2024.2386370
- Romano M, Arici ZS, Piskin D, et al. The 2021 EULAR/American College of Rheumatology points to consider for diagnosis, management and monitoring of the interleukin-1 mediated autoinflammatory diseases: cryopyrin-associated periodic syndromes, tumour necrosis factor receptor-associated periodic syndrome, mevalonate kinase deficiency, and deficiency of the interleukin-1 receptor antagonist. Ann Rheum Dis 2022; 81: 907-921. https://doi.org/10.1136/annrheumdis-2021-221801
- Koné-Paut I, Georgin-Lavialle S, Belot A, et al. Canakinumab treatment real world evidence in 3 monogenic periodic fever syndromes in 2009-2022: an interim analysis using the French JIR cohort database. Arthritis Res Ther 2024; 26: 80. https://doi.org/10.1186/s13075-024-03316-7
Copyright and license
Copyright © 2025 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









