
Graphical Abstract
Abstract
Background. Cystic fibrosis (CF) is a multisystem disease caused by variants in the CF transmembrane conductance regulator (CFTR) gene affecting ion transport. CFTR modulator therapy has become a significant treatment option for many CF patients. However, access to modulator therapy remains a challenge for cases with rare variants.
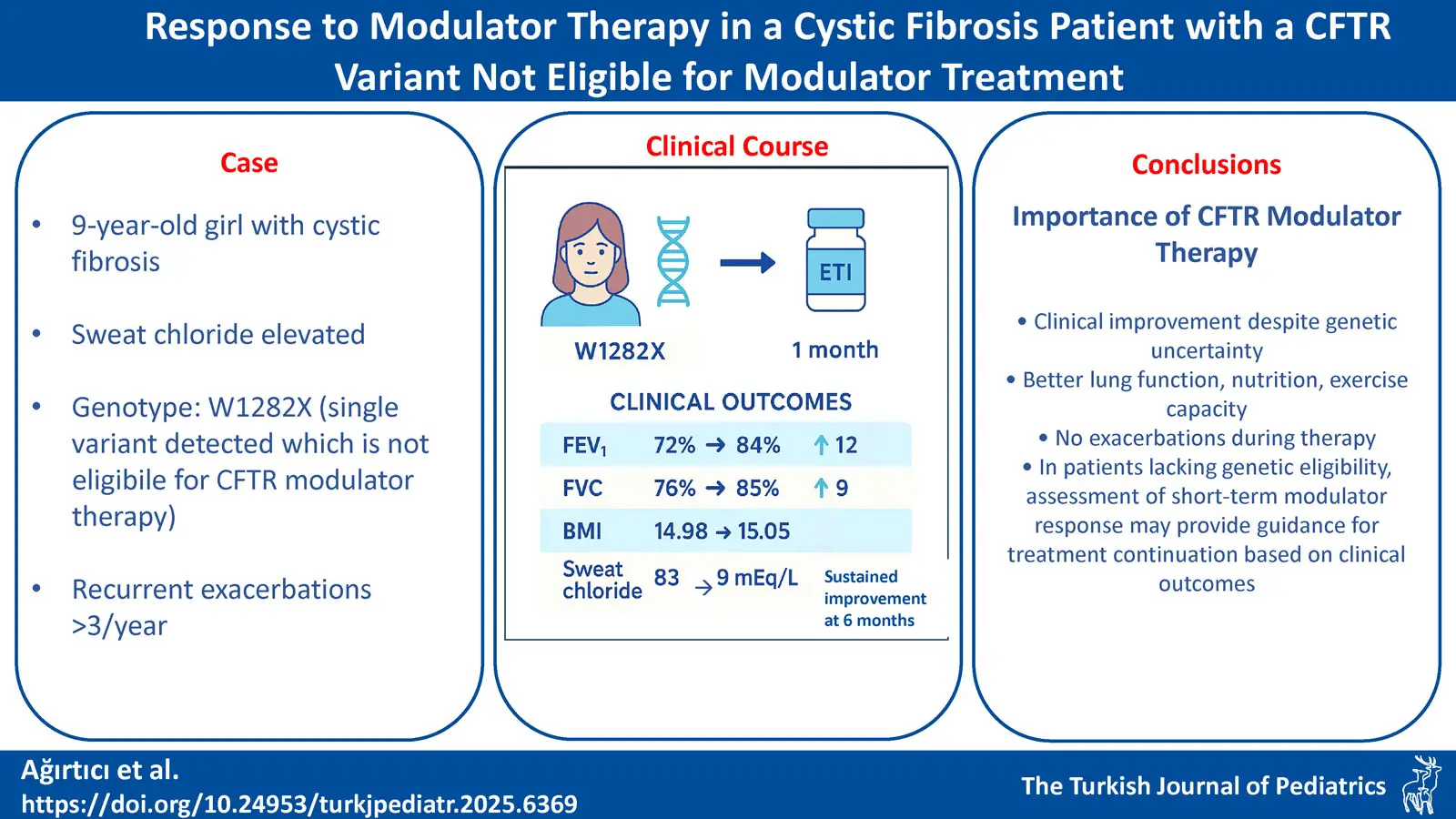
Case Presentation. Our 9-year-old female patient, diagnosed with CF by elevated sweat chloride and history of steatorrhea from birth, carried the rare W1282X variant with no clear eligibility for modulator therapy. The family self-financed one month of elexacaftor / tezacaftor / ivacaftor (ETI) treatment. After one-month, clinical evaluation showed improvements in body mass index (BMI; 14.98 to 15.05 kg/m2), an increase in forced expiratory volume in 1 second (FEV1%) by 12% (72% to 84%), decreased sweat chloride levels (from 83 mEq/L to 9 mEq/L), and enhanced exercise capacity. No pulmonary exacerbations occurred during therapy. Based on these findings, modulator therapy approval was obtained for continued treatment. Our patient is currently 10 years old and has been on modulator therapy for approximately 12 months.
Conclusions. Facilitating access to modulator therapies for patients with rare mutations is crucial, considering the potential long-term complications of CF. While organoid studies may not always predict clinical response, real-world cases demonstrate clinically meaningful benefit despite lack of organoid responsiveness. Short-term assessment of modulator response may not adequately reflect improvement in pulmonary function or exacerbation frequency, but decreases in sweat chloride, improvements in nutritional and functional parameters such as weight, BMI, and exercise capacity may be indicative of improvement in pulmonary function and exacerbation rates.
Keywords: cystic fibrosis, CFTR modulatory therapy, W1282X
Introduction
Cystic fibrosis (CF) is a monogenic disease caused by variants in the CF transmembrane conductance regulator (CFTR) gene, affecting chloride and water transport across epithelial cells, leading to thick, sticky secretions in multiple organs.1 The incidence of CF varies geographically, with 1 in 100,000–150,000 births in Asia and 1 in 3,500 in North America. CFTR mutations affect chloride channel function to varying degrees, resulting in a broad clinical spectrum. Respiratory manifestations include mucus plugging of small airways, air trapping, chronic productive cough, reduced spirometry volumes, recurrent lower respiratory infections, and bronchiectasis.2 Severe CF cases may become lung transplant candidates due to chronic inflammation, hypoxia, and infection. Recent advances in CF treatment, particularly CFTR modulators, have markedly improved disease management and quality of life by targeting underlying molecular defects.3 Ivacaftor monotherapy, through its potentiator effect, improves CFTR channel activity, resulting in better pulmonary function, reduced exacerbations, and weight gain in CF; however, its use is limited to ~5% of patients. The triple combination of elexacaftor / tezacaftor / ivacaftor (ETI) offering both corrector and potentiator actions, has demonstrated superior outcomes, particularly in those with one or two F508del alleles. Modulator eligibility is genotype-based, though for rare variants, responsiveness may be assessed in vitro using nasal epithelial samples or intestinal organoids in specialized centers.4
In Türkiye, the use of CFTR modulator therapy was officially authorized and included in the national reimbursement system as of July 2025. ETI combination therapy is reimbursed for patients aged ≥ 2 years with a confirmed diagnosis of CF (either by sweat chloride testing or genetic analysis) who carry at least one F508del mutation or another CFTR variant with proven responsiveness to the therapy. However, a subset of patients lacks identifiable mutations suitable for approved modulators, creating uncertainty regarding therapy benefits. In variants with premature termination codons, such as W1282X, modulator therapies are generally ineffective. Ongoing research for W1282X is exploring genetic approaches, including adenine base editing (ABE) and homology-independent targeted integration (HITI), as potential alternative treatment strategies.5 We present our experience with a patient clinically diagnosed with CF and a single identified CFTR variant (W1282X), who was not clearly eligible for modulators, but nonetheless demonstrated a positive response to ETI therapy.
Case Presentation
A 9-year-old girl diagnosed with CF at 2 months of age due to elevated sweat chloride (92.8 and 95 mEq/L) and history of steatorrhea was followed for bilateral bronchiectasis and chronic Pseudomonas aeruginosa infection. After a 12-month course of inhaled tobramycin, sputum cultures remained negative for P. aeruginosa. Pancreatic insufficiency was managed with enzyme replacement and fat-soluble vitamin supplementation; and ursodeoxycholic acid was given for CF-associated liver disease. She continued to receive dornase alfa and hypertonic saline inhalation, and chest physiotherapy, although not regularly. Whole exome sequencing (WES) and multiplex ligation-dependent probe amplification (MLPA) revealed a single W1282X variant on one allele of the CFTR gene, with no variant detected on the other allele. Non-adherence to chest physiotherapy and supportive treatments was associated with reduced exercise capacity and more than three pulmonary exacerbations annually. We were unable to identify our patient’s other variant with WES and MLPA. Ortiz et al. demonstrated ETI response in a case with W1282X and N1303K variant, which resulted in increased appetite, improved exercise capacity and body mass index (BMI), higher forced expiratory volume in 1 second (FEV1), and a reduced frequency of pulmonary exacerbations.6 Mutyam et al. observed clinical improvement in a cystic fibrosis patient who was homozygous for the W1282X mutation following treatment with ivacaftor; they attributed this to an increase in the activity of the residual protein.7 Given the existence of cases in the literature showing clinical benefit despite lack of clear genetic eligibility for CFTR modulatory treatment, a short-term trial of ETI therapy was planned. The patient’s family preferred to start ETI therapy despite uncertain eligibility. Informed consent was obtained from the family before treatment. Along with ETI, our patient continued concomitant treatment with dornase alfa, hypertonic saline, chest physiotherapy, and supportive therapies targeting other system involvements. At the initiation of treatment, she was able to maintain oxygen saturation on room air; however, sputum cultures revealed chronic methicillin-sensitive Staphylococcus aureus (MSSA) colonization. Computerized tomography (CT) of the chest demonstrated bronchial wall thickening and cylindrical bronchiectasis, as well as a mosaic attenuation pattern secondary to air trapping in the lungs. Baseline assessments included weight, spirometry, six-minute walk test and sweat chloride measurement.
Pre-treatment spirometry showed a forced vital capacity (FVC) of 76% and an FEV1 of 72%, which improved to 85% (+9%) and 84% (+12%), respectively, after one month of therapy, indicating marked improvement in lung volumes. The patient demonstrated improved exercise capacity, a substantial reduction in sweat chloride concentration (from 83 to 9 mEq/L) and a slight increase in BMI (from 14.98 to 15.05 kg/m²) (Table I). Although these short-term gains appear modest, they are clinically meaningful in pediatric CF, as they may contribute to catch-up growth, improved pulmonary function and a reduced frequency of exacerbations. No treatment-related adverse effects were observed.
| 6MWT: six-minute walking test; BMI: body mass index; FEF 25-75: forced expiratory flow between 25% and 75% of forced vital capacity; FVC: forced vital capacity; FEV1: forced expiratory volume in 1 second; FEV1/FVC: forced expiratory volume in 1 second to forced vital capacity ratio; PEF: peak expiratory flow. | |||
| Table I. Clinical and laboratory findings at the beginning, the first month and the sixth months of therapy. | |||
| Parameter |
|
|
|
| Weight |
|
|
|
| BMI |
(SDS: -0,77) |
(SDS: -0,89) |
(SDS: -0,83) |
| FVC |
|
|
|
| FEV1 |
|
|
|
| FEV1/FVC |
|
|
|
| FEF25-75 |
|
|
|
| PEF |
|
|
|
| Sweat chloride (mEq/L) |
|
|
|
| 6MWT |
|
|
|
| Number of pulmonary exacerbations |
|
|
|
Following these results, the family successfully obtained official approval for ongoing modulator therapy from the Ministry of Health. After six months of treatment, the patient’s growth parameters were 32 kg (SDS: -0,76), BMI 16.1 kg/m2, sputum cultures remained sterile, FEV₁ was 85% and no pulmonary exacerbations requiring hospitalization occurred (Table I).
Informed consent was obtained from the legal guardian for the use of the patient’s information in the publication.
Discussion
To our knowledge, this case represents the first reported patient in Türkiye with a single CFTR variant and no clear modulator eligibility, who responded well to ETI treatment. In our patient, the detection of only a single pathogenic variant, with the second variant not identified by MLPA or WES, precluded the initiation of modulator therapy. We presume that the treatment response may be attributable to the unidentified variant; however, as this could not be genetically confirmed, a formal eligibility assessment for the therapy could not be performed.
Due to the premature termination codon, CF patients harboring the W1282X mutation typically exhibit severe disease, with early chronic infections, pancreatic insufficiency, poor growth, impaired lung function, and early bronchiectasis.8
CFTR modulator drugs are classified as potentiators or correctors; potentiators enhance channel opening, and correctors improve cellular processing and trafficking.3 Clinical trials have demonstrated significant benefits of ETI in BMI, lung function, sweat chloride reduction, and decreased infections.9,10 However, not all patients harbor mutations eligible for modulator therapy. A recent Turkish national registry study found that 57.2% of CF patients were eligible for modulators.11
For patients without clearly identified variants, organoid assays may help predict modulator responsiveness.12 Unfortunately, such testing is not yet available in Türkiye. Experimental studies in mice suggest variable responses to modulators among rare mutations, with W1282X showing relatively better response than others.13
Premature termination codon mutations, such as W1282X, currently lack effective RNA-targeted therapies.14 Nonsense-mediated mRNA decay (NMD) further limits the response to CFTR modulators by degrading the truncated protein. Experimental approaches have demonstrated that selective inhibition of NMD can partially restore protein function when combined with modulator therapy, offering a potential adjunctive strategy; however, due to the essential roles of NMD in normal cellular processes, precise targeting is required.8,15
In their experimental study, Haggie et al. demonstrated that treatment responsiveness could be achieved in W1282X variants using one corrector and two potentiators, and suggested that the currently available triple combination, containing two correctors and one potentiator, may be suboptimal in such cases.16 In our patient, although the specific variant typically associated with responsiveness to the standard combination was not identified, a clear clinical benefit was achieved with the two-corrector/one-potentiator regimen. We believe that this response may be attributable to an unidentified second pathogenic variant. Clinical data also support modulator benefits in patients with rare or unidentified variants, even when organoid results are inconclusive.17 Thus, clinical response remains the most reliable indicator for continuing treatment.
Conclusion
CF is a multisystem disease and genetic characterization is essential for modulator therapy eligibility, but some patients remain without identified variants. Early initiation of modulator therapy is vital for improving quality of life and preventing complications. New pathogenic variants are increasingly being recognized. In patients lacking genetic eligibility in whom organoid studies cannot be performed, assessment of short-term modulator response may provide guidance for treatment continuation based on clinical outcomes. Treatment options aimed at improving quality of life in chronic diseases should always be thoroughly evaluated, with individualized consideration given to each patient’s unique circumstances.
Ethical approval
Informed consent was obtained from the legal guardian for the use of the patient’s information in the publication.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Bush A. Learning from cystic fibrosis: how can we start to personalise treatment of Children’s Interstitial Lung Disease (chILD)? Paediatr Respir Rev 2024; 50: 46-53. https://doi.org/10.1016/j.prrv.2023.11.001
- Turcios NL. Cystic fibrosis lung disease: an overview. Respir Care 2020; 65: 233-251. https://doi.org/10.4187/respcare.06697
- Corrao F, Kelly-Aubert M, Sermet-Gaudelus I, Semeraro M. Unmet challenges in cystic fibrosis treatment with modulators. Expert Rev Respir Med 2024; 18: 145-157. https://doi.org/10.1080/17476348.2024.2357210
- Grasemann H, Ratjen F. Cystic fibrosis. N Engl J Med 2023; 389: 1693-1707. https://doi.org/10.1056/NEJMra2216474
- Mention K, Cavusoglu-Doran K, Joynt AT, et al. Use of adenine base editing and homology-independent targeted integration strategies to correct the cystic fibrosis causing variant, W1282X. Hum Mol Genet 2023; 32: 3237-3248. https://doi.org/10.1093/hmg/ddad143
- Tupayachi Ortiz MG, Baumlin N, Yoshida M, Salathe M. Response to Elexacaftor/Tezacaftor/Ivacaftor in people with cystic fibrosis with the N1303K mutation: Case report and review of the literature. Heliyon 2024; 10: e26955. https://doi.org/10.1016/j.heliyon.2024.e26955
- Mutyam V, Libby EF, Peng N, et al. Therapeutic benefit observed with the CFTR potentiator, ivacaftor, in a CF patient homozygous for the W1282X CFTR nonsense mutation. J Cyst Fibros 2017; 16: 24-29. https://doi.org/10.1016/j.jcf.2016.09.005
- Kim YJ, Nomakuchi T, Papaleonidopoulou F, Yang L, Zhang Q, Krainer AR. Gene-specific nonsense-mediated mRNA decay targeting for cystic fibrosis therapy. Nat Commun 2022; 13: 2978. https://doi.org/10.1038/s41467-022-30668-y
- Miller AC, Harris LM, Cavanaugh JE, et al. The rapid reduction of infection-related visits and antibiotic use among people with cystic fibrosis after starting elexacaftor-tezacaftor-ivacaftor. Clin Infect Dis 2022; 75: 1115-1122. https://doi.org/10.1093/cid/ciac117
- Migliorisi G, Collura M, Ficili F, et al. Elexacaftor-tezacaftor-ivacaftor as a final frontier in the treatment of cystic fibrosis: definition of the clinical and microbiological implications in a case-control study. Pharmaceuticals (Basel) 2022; 15: 606. https://doi.org/10.3390/ph15050606
- Nayır Büyükşahin H, Emiralioğlu N, Yalçın E, et al. Comparison of clinical features of cystic fibrosis patients eligible but not on CFTR modulators to ineligible for CFTR modulators. Pediatr Pulmonol 2024; 59: 2499-2506. https://doi.org/10.1002/ppul.27051
- Michicich M, Traylor Z, McCoy C, et al. A W1282X cystic fibrosis mouse allows the study of pharmacological and gene-editing therapeutics to restore CFTR function. J Cyst Fibros 2025; 24: 164-174. https://doi.org/10.1016/j.jcf.2024.10.008
- Comegna M, Terlizzi V, Salvatore D, et al. Elexacaftor-tezacaftor-ivacaftor therapy for cystic fibrosis patients with the F508del/unknown genotype. Antibiotics (Basel) 2021; 10: 828. https://doi.org/10.3390/antibiotics10070828
- McHugh DR, Steele MS, Valerio DM, et al. A G542X cystic fibrosis mouse model for examining nonsense mutation directed therapies. PLoS One 2018; 13: e0199573. https://doi.org/10.1371/journal.pone.0199573
- Aksit MA, Bowling AD, Evans TA, et al. Decreased mRNA and protein stability of W1282X limits response to modulator therapy. J Cyst Fibros 2019; 18: 606-613. https://doi.org/10.1016/j.jcf.2019.02.009
- Haggie PM, Phuan PW, Tan JA, et al. Correctors and potentiators rescue function of the truncated W1282X-cystic fibrosis transmembrane regulator (CFTR) translation product. J Biol Chem 2017; 292: 771-785. https://doi.org/10.1074/jbc.M116.764720
- Fainardi V, Cresta F, Sorio C, et al. Elexacaftor/tezacaftor/ivacaftor in people with cystic fibrosis and rare mutations. Pediatr Pulmonol 2024; 59: 3383-3390. https://doi.org/10.1002/ppul.27211
Copyright and license
Copyright © 2025 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









