
Graphical Abstract
Abstract
Background. Leber hereditary optic neuropathy (LHON) is a maternally inherited mitochondrial disorder that predominantly manifests as bilateral, painless vision loss in young males. While traditionally associated with the optic nerves, a subset of patients exhibits additional neurological symptoms, referred to as LHON-plus syndrome. Involvement of the spinal cord is uncommon, particularly among the pediatric population, and may result in diagnostic challenges, potentially leading to confusion regarding acquired demyelinating diseases.
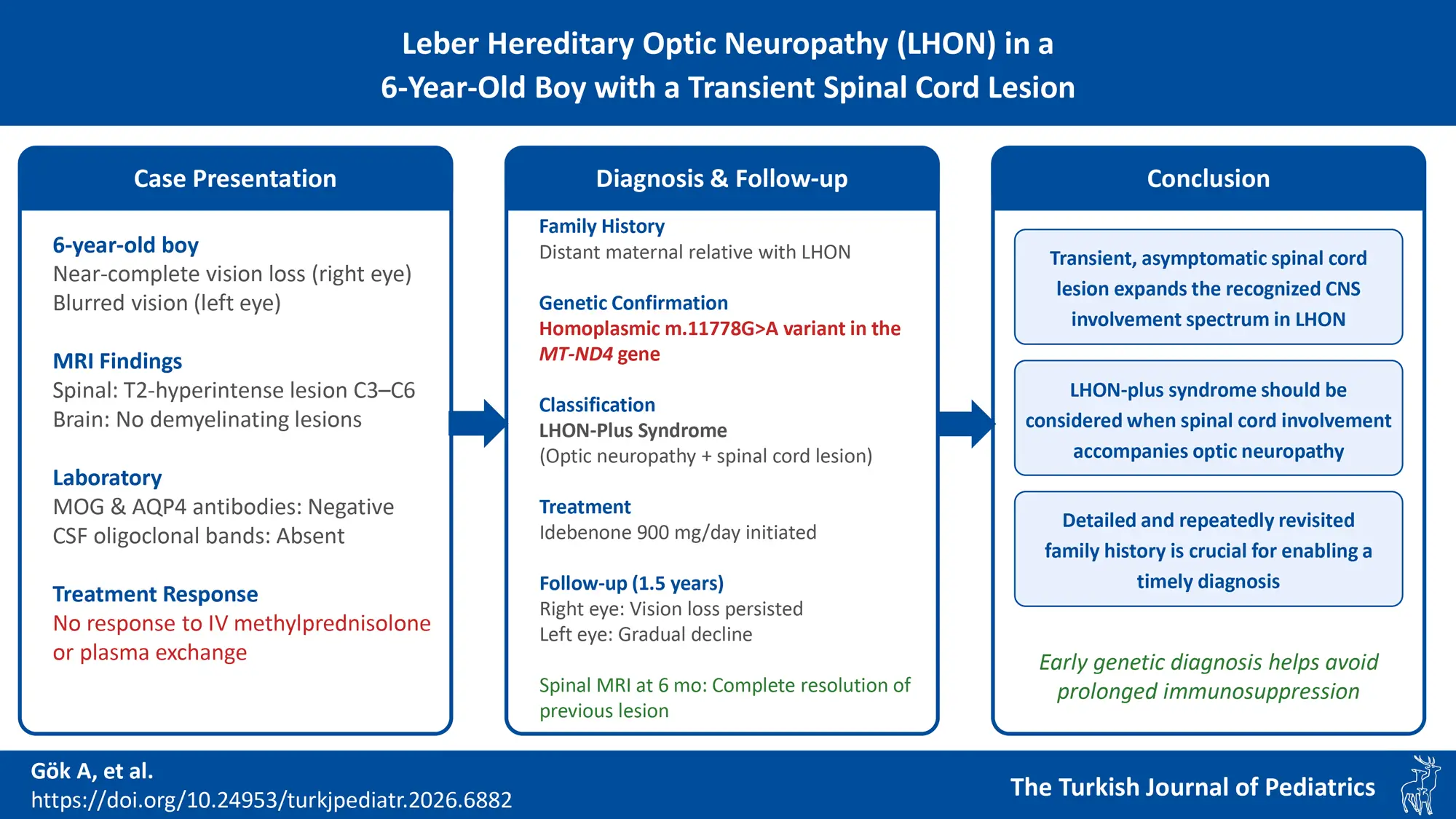
Case Presentation. We report a 6-year-old boy with near-complete vision loss in the right eye and blurred vision in the left eye. Demyelinating diseases were suspected in the case of acute bilateral optic neuropathy. Cranial and orbital magnetic resonance imaging (MRI) showed no demyelinating lesions, whereas spinal MRI revealed a T2-hyperintense lesion at the C3–C6 levels. Due to unresponsiveness to conventional treatment for demyelinating diseases, genetic testing confirmed the homoplasmic m.11778G>A variant in NADH dehydrogenase subunit 4 (MT-ND4), establishing an LHON diagnosis. Spinal cord involvement supported the LHON-plus syndrome classification. Idebenone therapy was initiated, and follow-up was scheduled. During the 1.5-year follow-up, right eye visual loss persisted, while the left eye showed gradual vision decline. A second spinal MRI performed at 6 months showed complete resolution of the previous lesion without new lesions.
Conclusion. Since optic neuropathy and spinal cord involvement typically indicate demyelinating diseases, these should be prioritized in initial evaluation due to their frequency and need for early immunomodulatory treatment. However, spinal cord involvement can occur in mitochondrial diseases, as demonstrated in this case. The presence of a transient and asymptomatic spinal cord lesion in this patient expands the recognized spectrum of central nervous system involvement in LHON. Therefore, LHON-plus syndrome should be considered when spinal cord involvement accompanies optic neuropathy after excluding other demyelinating diseases.
Keywords: Leber hereditary optic neuropathy, LHON-plus syndrome, optic neuropathy, mitochondrial disease, MT-ND4 m.11778G>A mutation
Introduction
Acute or subacute vision loss in children may result from various causes, including demyelinating optic neuritis, increased intracranial pressure, toxic or metabolic optic neuropathy, mitochondrial disorders and hereditary optic neuropathy. However, because children may have difficulty accurately describing visual symptoms, some cases that appear to present with acute vision loss may represent a slowly progressive or chronic decline in vision that has only recently become evident.1
Leber hereditary optic neuropathy (LHON) is a genetic disorder that is transmitted through maternal inheritance. A mutation in the mitochondrial DNA (mtDNA) causes this disorder, leading to the degeneration of retinal ganglion cells and the subsequent optic nerve. LHON manifests as a gradual, painless loss of central vision in both eyes, primarily affecting young adult males.2 Although LHON predominantly affects young males, homoplasmic males are at particularly high risk of developing visual loss, whereas disease penetrance is substantially lower in females, even among homoplasmic carriers.2 While LHON is generally confined to the optic nerves, a subset of patients present with additional neurological and non-neurological manifestations, a condition known as the LHON-plus syndrome. Extraocular manifestations have been documented, including cardiac arrhythmias, skeletal changes, myopathy, dementia, movement disorders, peripheral neuropathy, brainstem and basal ganglia involvement and multiple sclerosis (MS)-like syndromes. Such extraocular manifestations complicate the clinical presentation and pose diagnostic challenges.3,4
Herein, we report the detection of spinal cord involvement in a 6-year-old boy who presented with optic neuropathy and was subsequently diagnosed with LHON following comprehensive investigations. We underscore that LHON should be considered as a differential diagnosis for optic neuropathy, even in the presence of spinal cord lesions.
Case Presentation
A 6-year-old boy presented with a one-week history of difficulty focusing on objects and a deviated gaze, as noted by his mother. She also reported that he had been watching television from a closer distance over the previous month, and recent photographs showed an inability to focus on the camera. Initial ophthalmological examination revealed a visual acuity of 0.1 in the right eye, consistent with severe visual impairment, and subjective blurred vision in the left eye, despite normal visual acuity. Bilateral papilledema was observed during fundoscopic examination. Based on these findings, the patient was referred to the pediatric neurology department for further evaluation.
On comprehensive ophthalmological examination, the right optic disc exhibited blurred margins, whereas the left optic disc showed swelling consistent with papilledema. The right eye demonstrated diminished light reflexes, and optical coherence tomography (OCT) revealed retinal nerve fiber layer (RNFL) thickening in the left eye and thinning in the right eye, reflecting different stages of optic neuropathy. Despite these ocular findings, the patient exhibited no additional neurological deficits on physical examination, and motor and sensory functions remained intact.
At initial presentation, there was no known family history of visual loss or hereditary optic neuropathy. The family history was specifically questioned and was unremarkable at that time. The patient was also evaluated early by an experienced neuro-ophthalmologist, who did not consider hereditary optic neuropathy as the primary diagnosis based on the clinical findings.
Given the severity of visual impairment and absence of other neurological signs, the patient underwent advanced imaging studies. Spinal and brain magnetic resonance imaging (MRI) was taken to differentiate demyelinating optic neuropathies (myelin oligodendrocyte glycoprotein antibody disease - MOGAD, neuromyelitis optica – NMO, and MS) in the differential diagnosis of optic neuropathy. Spinal MRI revealed diffuse intramedullary increased signal intensity in the cervical spinal cord at the C3–C6 levels on T2-weighted images; however, no associated cord edema was observed (Fig. 1). Furthermore, brain imaging demonstrated no evidence of demyelination in T2 and fluid-attenuated inversion recovery (FLAIR) sequences (Fig. 2).
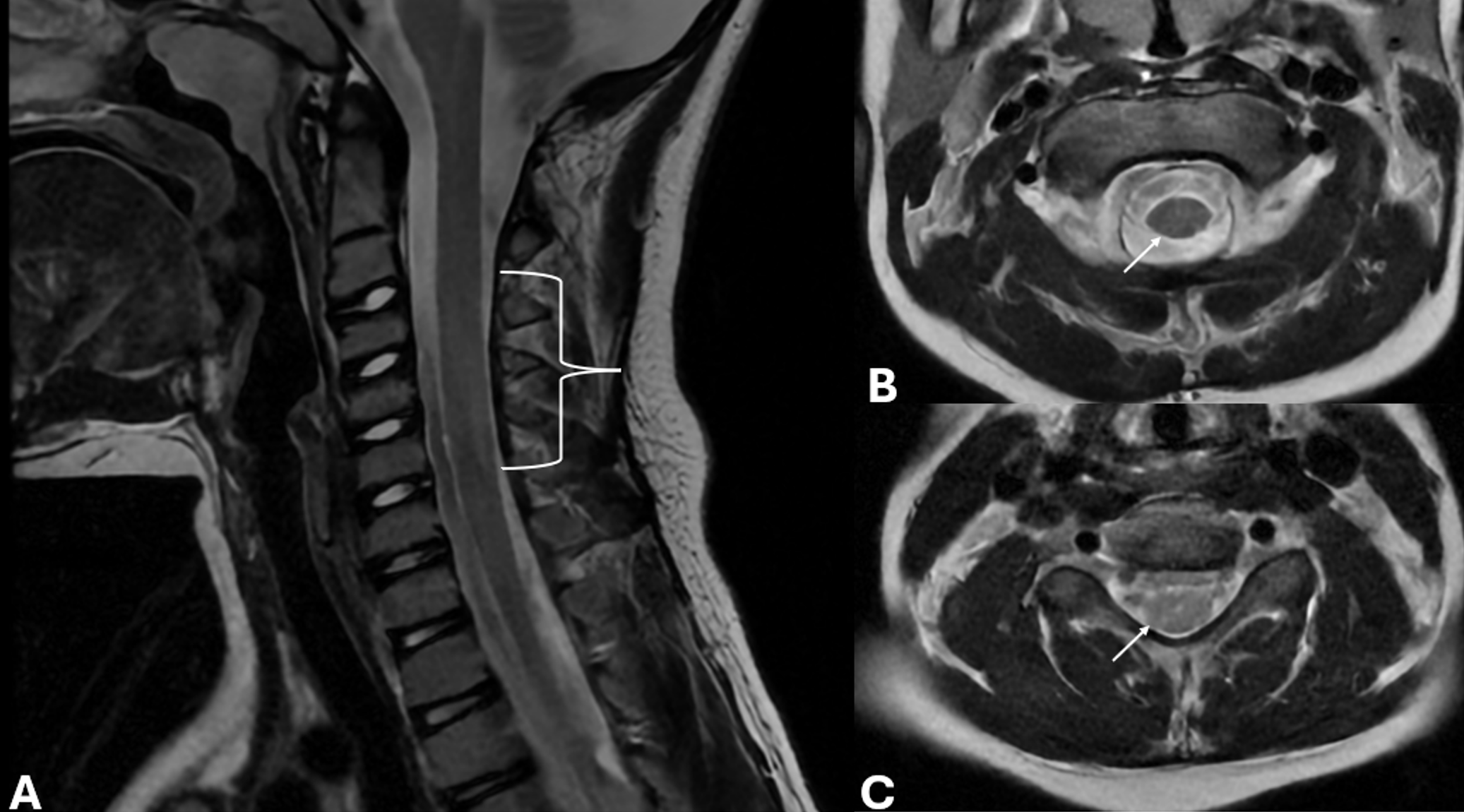
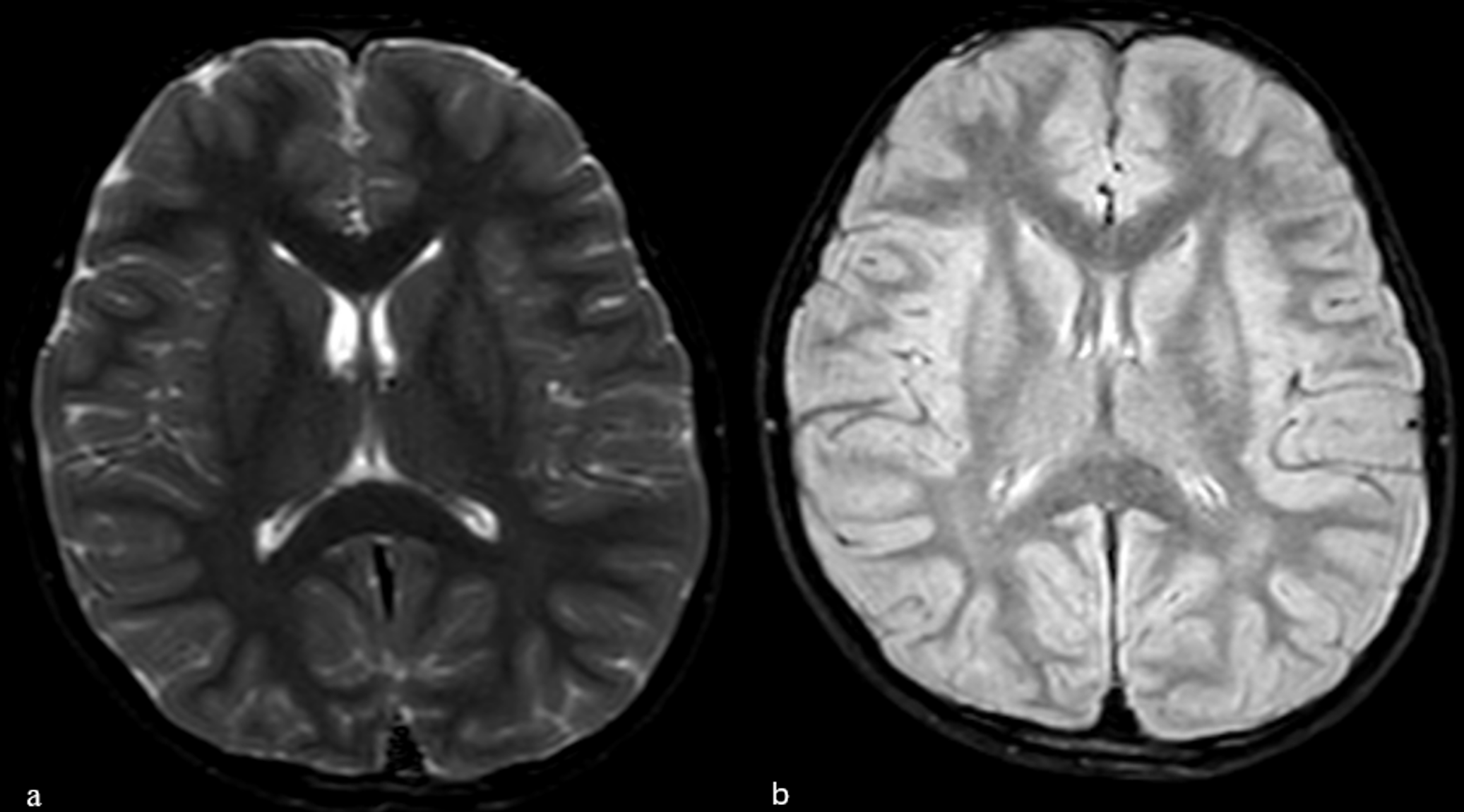
The MRI findings, combined with ophthalmologic examination, prompted further investigation into potential demyelinating optic neuropathies. These were ultimately ruled out through cerebrospinal fluid (CSF) analysis and antibody testing. Both serum myelin oligodendrocyte glycoprotein (MOG) and aquaporin-4 (AQP4) antibodies were negative and CSF oligoclonal bands were absent, and the IgG index was within the normal range.
Pending the availability of results, the patient was initiated on therapy for autoimmune and demyelinating diseases and the therapeutic response was subsequently monitored. Initial management consisted of high-dose intravenous methylprednisolone administration at 30 mg/kg/day for five days. Due to the lack of clinical improvement, the steroid course was extended to a total of seven days; however, no significant response was observed. Consequently, the patient underwent seven courses of daily plasma exchange therapy, again without substantial clinical improvement.
During the prolonged hospital stay, increased intra-family communication led distant relatives to become aware of the patient’s condition. As a result, a family member with a known diagnosis of LHON directly contacted the patient’s family. This information emerged after spinal cord involvement had been identified and plasma exchange therapy had already been initiated. The newly obtained family history subsequently prompted reconsideration of the diagnosis and targeted genetic evaluation.
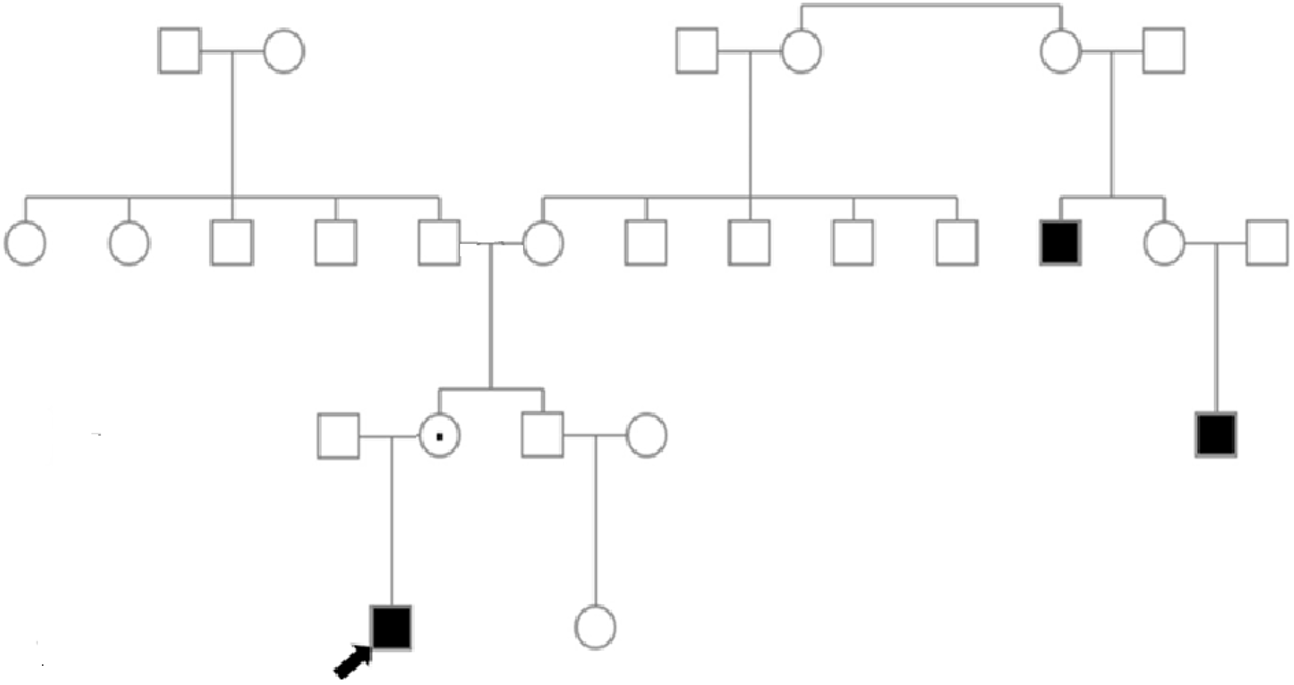
Given the family history of a distant maternal relative who was previously diagnosed with LHON, genetic testing was conducted to confirm the diagnosis. Genetic analysis revealed the same homoplasmic m.11778G>A variant in the NADH dehydrogenase subunit 4 (MT-ND4) gene, which is strongly associated with LHON. Genetic analysis of the patient’s mother revealed the same homoplasmic variant. Mitochondrial DNA testing was conducted on peripheral blood samples collected from the patient and his mother. The mother, who was asymptomatic, was closely monitored as an asymptomatic carrier. Fig. 3 illustrates the pedigree. Detailed genetic counseling regarding LHON was provided to the mother and other family members at risk.
The diagnosis of LHON was genetically confirmed, and spinal involvement was classified as LHON-plus syndrome. Idebenone therapy was initiated at a daily dose of 900 mg, and the patient was followed up for changes in visual acuity and overall clinical course.
During follow-up, no improvement was observed in the right eye, whereas the left eye showed a gradual decline in visual acuity over time. However, due to the variable visual performance across different visual field regions, the ophthalmologist did not report a single consistent visual acuity value for the left eye. A follow-up spinal MRI performed six months later demonstrated complete resolution of the previous lesion.
Written informed consent was obtained from the patient’s parents for publication of this case report and accompanying images.
Discussion
Acute optic neuropathy in children represents a diagnostic challenge due to its broad etiological spectrum of causes. A thorough history, ophthalmological examination, and neuroimaging are essential for narrowing the differential diagnosis. In the initial evaluation, it is important for clinicians, especially pediatricians, to first consider demyelinating disorders, given their higher prevalence and the critical importance of early immunomodulatory therapy. MRI of the brain and orbits is typically indicated within the first few days of presentation to identify demyelinating or compressive causes of the symptoms. Spinal MRI is usually not a routine part of the initial evaluation in children without signs of spinal cord dysfunction; however, it may be warranted when optic neuropathy is accompanied by atypical imaging findings or when demyelinating diseases are considered. Lumbar puncture and CSF analysis are valuable for excluding inflammatory or infectious etiologies and may reveal demyelinating markers.
Although LHON most commonly manifests in young adulthood, pediatric-onset cases are rare; therefore, the onset of symptoms at 6 years of age represents an unusual and noteworthy aspect of this case.
LHON is traditionally characterized as an isolated optic neuropathy. However, a subset of patients exhibit additional neurological and non-neurological features collectively referred to as “LHON-plus”.5 The documented neurological manifestations of LHON-plus include movement disorders, peripheral neuropathy, dysfunction of the brainstem or basal ganglia, and demyelinating syndromes that resemble MS.6 In a case series involving patients with LHON (n = 46), 59% (n = 27) demonstrated neurological or extraocular symptoms.2 To our knowledge, spinal cord involvement in children with LHON has only been reported in isolated cases. For instance, Bursle et al.5 described an 8-year-old boy with dual mitochondrial DNA mutations at positions 14484 (MT-ND6) and 4160 (MT-ND1) who developed longitudinally extensive transverse myelitis (LETM). Another recent report described a 2-year-old girl who was initially diagnosed with antibody-negative NMO spectrum disorder (NMOSD) before genetic testing revealed a LHON mutation (m.14484T>C), subsequently diagnosed as LHON-plus.6 This LHON-plus case was initially treated as a demyelinating disease for two years until the mitochondrial mutation was identified. In our patient, early genetic diagnosis helped avoid prolonged immunosuppression and allowed appropriate management (initiation of idebenone) and genetic counseling for the family. Despite the rarity of both LHON and NMO, there have been documented instances of individuals possessing both the pathogenic mtDNA sequence variants associated with LHON and anti-AQP4 seropositive NMOSD.7-9 McClelland et al.10 described a 65-year-old woman with LHON who had been masquerading as NMOSD, and they emphasized that LHON should be considered in any case of bilateral or atypical optic neuritis, even in presumed seronegative NMO.
Mitochondrial disorders are increasingly being recognized as multisystem diseases that can affect the spinal cord, resulting in a diverse range of neurological manifestations. The identification of spinal cord involvement is essential for appropriate management.11 In a 2021 study by Alves et al.12, spinal MRI scans of 33 children with genetically confirmed primary mitochondrial disorders were retrospectively analyzed. Notably, 19 (58 %) patients had spinal cord lesions. These lesions were classified into two radiological patterns: Group A (n=12) displayed white ± gray matter involvement, resembling acquired demyelinating disorders, such as neuromyelitis optica spectrum disorders, multiple sclerosis, or MOG-antibody-associated disease; Group B (n=7) showed isolated gray matter involvement, mimicking ischemic or infectious etiologies. These findings highlight the diversity of spinal cord presentations in MIDs and their potential for misdiagnoses.
The limited visual recovery observed in our patient despite idebenone therapy may be explained by the underlying genetic variant and the rapid disease course. The m.11778G>A variant is known to be associated with the poorest visual prognosis among the common LHON mutations. In addition, rapid progression of visual symptoms, as observed within one week in this case, has been reported as an unfavorable prognostic factor for visual recovery.13
In cases where optic neuropathy is accompanied by spinal cord lesions, demyelinating diseases should be primarily considered, as spinal involvement is not typical of LHON. The presence of a transient spinal cord lesion without clinical symptoms appears to be a rare and potentially novel finding in LHON, broadening the recognized spectrum of central nervous system involvement.
Pediatricians, pediatric neurologists, and eye care professionals should include LHON in their differential diagnoses when evaluating patients with unexplained optic neuropathy even when additional neurological symptoms or imaging findings are present. From a general pediatrics perspective, this case highlights the critical importance of a detailed family history, which ultimately played a key role in establishing the diagnosis and redirecting the diagnostic approach. Even if this had been the first affected individual in the family without a known family history, the treatment-resistant and atypical clinical course would have prompted consideration of genetic testing for hereditary optic neuropathies, but at a later stage. A detailed and repeatedly revisited family history played a crucial role in enabling a more timely diagnosis in this case.
Ethical approval
Written informed consent was obtained from the patient’s parents for publication of this case report and accompanying images.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Iqbal S, Klein BL. Diagnostic approach to acute vision loss in children. In: Teach SJ, Wiley JF, editors. UpToDate. 2025. Available at: https://www.uptodate.com/contents/diagnostic-approach-to-acute-vision-loss-in-children
- Mauri E, Dilena R, Boccazzi A, et al. Subclinical Leber’s hereditary optic neuropathy with pediatric acute spinal cord onset: more than meets the eye. BMC Neurol 2018; 18: 220. https://doi.org/10.1186/s12883-018-1227-9
- Nikoskelainen EK, Marttila RJ, Huoponen K, et al. Leber’s “plus”: neurological abnormalities in patients with Leber’s hereditary optic neuropathy. J Neurol Neurosurg Psychiatry 1995; 59: 160-164. https://doi.org/10.1136/jnnp.59.2.160
- Sun MM, Zhou HF, Sun Q, et al. Leber’s hereditary optic neuropathy companied with multiple-related diseases. Front Hum Neurosci 2022; 16: 964550. https://doi.org/10.3389/fnhum.2022.964550
- Bursle C, Riney K, Stringer J, et al. Leber hereditary optic neuropathy and longitudinally extensive transverse myelitis. JIMD Rep 2018; 42: 53-60. https://doi.org/10.1007/8904_2017_79
- Sunshine A, Mandle QJ, Cabal Herrera AM, Zapanta B, Varma H, Magaña S. Pearls & Oy-sters: Leber hereditary optic neuropathy-plus masquerading as neuromyelitis optica spectrum disorder in a 2-year-old child. Neurology 2023; 101: e2585-e2588. https://doi.org/10.1212/WNL.0000000000207979
- Shiraishi W, Hayashi S, Kamada T, et al. A case of neuromyelitis optica harboring both anti-aquaporin-4 antibodies and a pathogenic mitochondrial DNA mutation for Leber’s hereditary optic neuropathy. Mult Scler 2014; 20: 258-260. https://doi.org/10.1177/1352458513513057
- Dujmovic I, Jancic J, Dobricic V, et al. Are Leber’s mitochondial DNA mutations associated with aquaporin-4 autoimmunity? Mult Scler 2016; 22: 393-394. https://doi.org/10.1177/1352458515590649
- Simão LM. Neuromyelitis optica antibody in Leber hereditary optic neuropathy: case report. Arq Bras Oftalmol 2012; 75: 280-282. https://doi.org/10.1590/s0004-27492012000400013
- McClelland CM, Van Stavern GP, Tselis AC. Leber hereditary optic neuropathy mimicking neuromyelitis optica. J Neuroophthalmol 2011; 31: 265-268. https://doi.org/10.1097/WNO.0b013e318225247b
- Finsterer J, Zarrouk-Mahjoub S. Involvement of the spinal cord in mitochondrial disorders. J Neurosci Rural Pract 2018; 9: 245-251. https://doi.org/10.4103/jnrp.jnrp_446_17
- Alves CAPF, Goldstein A, Teixeira SR, et al. Involvement of the spinal cord in primary mitochondrial disorders: a neuroimaging mimicker of inflammation and ischemia in children. AJNR Am J Neuroradiol 2021; 42: 389-396. https://doi.org/10.3174/ajnr.A6910
- Newman NJ, Carelli V, Taiel M, Yu-Wai-Man P. Visual outcomes in Leber hereditary optic neuropathy patients with the m.11778G>A (MTND4) mitochondrial DNA mutation. J Neuroophthalmol 2020; 40: 547-557. https://doi.org/10.1097/WNO.0000000000001045
Copyright and license
Copyright © 2026 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









