
Abstract
Background. Mevalonate kinase deficiency (MKD) is a rare autosomal recessive autoinflammatory disease. Chronic nonbacterial osteomyelitis (CNO) represents another autoinflammatory disorder characterized by sterile bone inflammation. Although musculoskeletal pain is common in MKD, CNO-like bone lesions have rarely been described. Tocilizumab has been used separately in MKD / hyper-IgD syndrome (HIDS) and in CNO; however, pediatric cases showing both conditions together and responding completely to IL-6 blockade have not been previously reported.
Case Presentation. We present a 16-year-old boy born to consanguineous parents who experienced early-onset recurrent fever episodes with abdominal pain, maculopapular rash, oral ulcers, and conjunctival injection. He was initially misdiagnosed with FMF and IgA vasculitis and treated with colchicine with partial improvement. Persistent systemic inflammation prompted referral to our tertiary center at age 9. A periodic fever gene panel revealed a homozygous MVK p.V377I mutation, confirming MKD.
Anakinra therapy was initiated. Although no attacks occurred during the first month, flares characterized by fever, abdominal pain, rash, and elevated acute-phase reactants recurred in the second and third months, reflecting a partial response. Consequently, treatment was switched to canakinumab. Toward the end of the second year of IL-1 blockade, attack frequency increased to once every three months, and at age 12 he developed diffuse musculoskeletal pain. Whole-body magnetic resonance imaging demonstrated multifocal metaphyseal bone marrow edema compatible with CNO-like lesions. Sulfasalazine was added but discontinued after drug-induced pancreatitis. Given persistent systemic inflammation and inadequate response to IL-1 inhibitors, canakinumab was replaced with weekly subcutaneous tocilizumab, resulting in rapid improvement in bone pain and systemic inflammation. Since the third month of tocilizumab therapy, he has remained clinically stable with normalized inflammatory markers and only a single mild flare over the last year.
Conclusion. This case highlights the potential role of IL-6 inhibition in complicated MKD presenting with autoinflammatory bone disease refractory to standard therapies.
Keywords: mevalonate kinase deficiency, hyper-IgD syndrome, chronic nonbacterial osteomyelitis, autoinflammatory bone disease, tocilizumab
Introduction
Mevalonate kinase deficiency (MKD) is a rare autosomal recessive autoinflammatory disease that encompasses a clinical spectrum ranging from the milder hyperimmunoglobulin D syndrome (HIDS) to the more severe mevalonic aciduria.1,2 MKD typically manifests with early-onset recurrent febrile episodes accompanied by cervical lymphadenopathy, abdominal pain, diarrhea, and aphthous ulcers.1,2 The disorder is caused by biallelic mutations in the MVK gene, with the p.V377I variant being the most frequently identified mutation associated with the HIDS phenotype.2-5
Chronic nonbacterial osteomyelitis (CNO) represents another autoinflammatory disorder characterized by sterile, relapsing-remitting bone inflammation mediated by dysregulated interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and IL-6 pathways.6-9 While musculoskeletal manifestations such as arthralgia and transient arthritis are commonly observed during MKD flares, radiologically confirmed CNO-like bone lesions have only rarely been described in the context of monogenic autoinflammatory diseases.9,10
IL-1 inhibitors are the primary biologic therapy in MKD; however, refractory cases exist.2,11-13 Alternative cytokine pathways, including IL-6, play important roles in systemic inflammation and bone remodeling.8-10 Tocilizumab, a monoclonal antibody targeting the IL-6 receptor, has been utilized independently in the management of both MKD/HIDS and CNO.12-15
To date, pediatric cases presenting with a coexistence of both conditions, particularly those demonstrating a complete clinical and radiological response to IL-6 blockade after failing IL-1 inhibition, have not been previously reported. Herein we present this case to underscore the diagnostic and therapeutic challenges involved and to highlight the potential role of the IL-6 pathway in refractory musculoskeletal involvement of MKD.
Case Presentation
A 16-year-old boy, born to consanguineous parents (first-degree cousins), had recurrent fever episodes beginning at 4 months of age. Attacks were associated with abdominal pain, maculopapular rash, oral ulcers, and conjunctival injection. He was initially followed with a clinical suspicion of familial Mediterranean fever (FMF) and IgA vasculitis at another clinic. MEFV gene analysis was performed during this period and showed no pathogenic mutations; however, he was treated with colchicine with partial improvement due to the symptomatic overlap.
At age 9, he was referred to our center due to persistent systemic inflammation despite treatment. He had a history of spontaneous epistaxis and isolated thrombocytopenia at age 8. Anti-nuclear antibody (ANA) testing was positive at 2+ intensity with a granular pattern. Given the ANA positivity and history of thrombocytopenia, further evaluation was performed to rule out systemic lupus erythematosus (SLE). Although the extractable nuclear antigen (ENA) panel showed isolated anti-DFS70 positivity, this was considered clinically insignificant. Anti-dsDNA was negative and complement levels were normal. Ultimately, the absence of specific clinical features and a normal urinalysis supported the exclusion of SLE.
A periodic fever gene panel (MEFV, ADA2, MVK, NLRP3, NLRP12, TNFRSF1A, TNFRSF11A, LPIN2, PSTPIP1, IL1RN, IL10RA, IL10RB, and NOD2) identified a homozygous MVK p.V377I mutation, confirming MKD with a HIDS phenotype.
Anakinra 100 mg/day (3 mg/kg/day) was initiated. After approximately three months of therapy, no attacks occurred during the first month; however, in the second and third months he experienced flares lasting about seven days, characterized by fever, abdominal pain, rash, and elevated acute-phase reactants. Therefore, his response to anakinra was considered partial, and treatment was switched to canakinumab 150 mg/month (4 mg/kg/month). Toward the end of the second year, the attack frequency increased to once every three months, and at age 12 he developed progressive, diffuse musculoskeletal pain while still receiving canakinumab. He had no morning stiffness or inflammatory back pain; however, there was marked bone tenderness on palpation, particularly in the clavicle, femur, tibia, and humerus. Whole-body magnetic resonance imaging (MRI) revealed multifocal metaphyseal bone marrow edema in these regions, compatible with CNO-like lesions (Fig. 1).
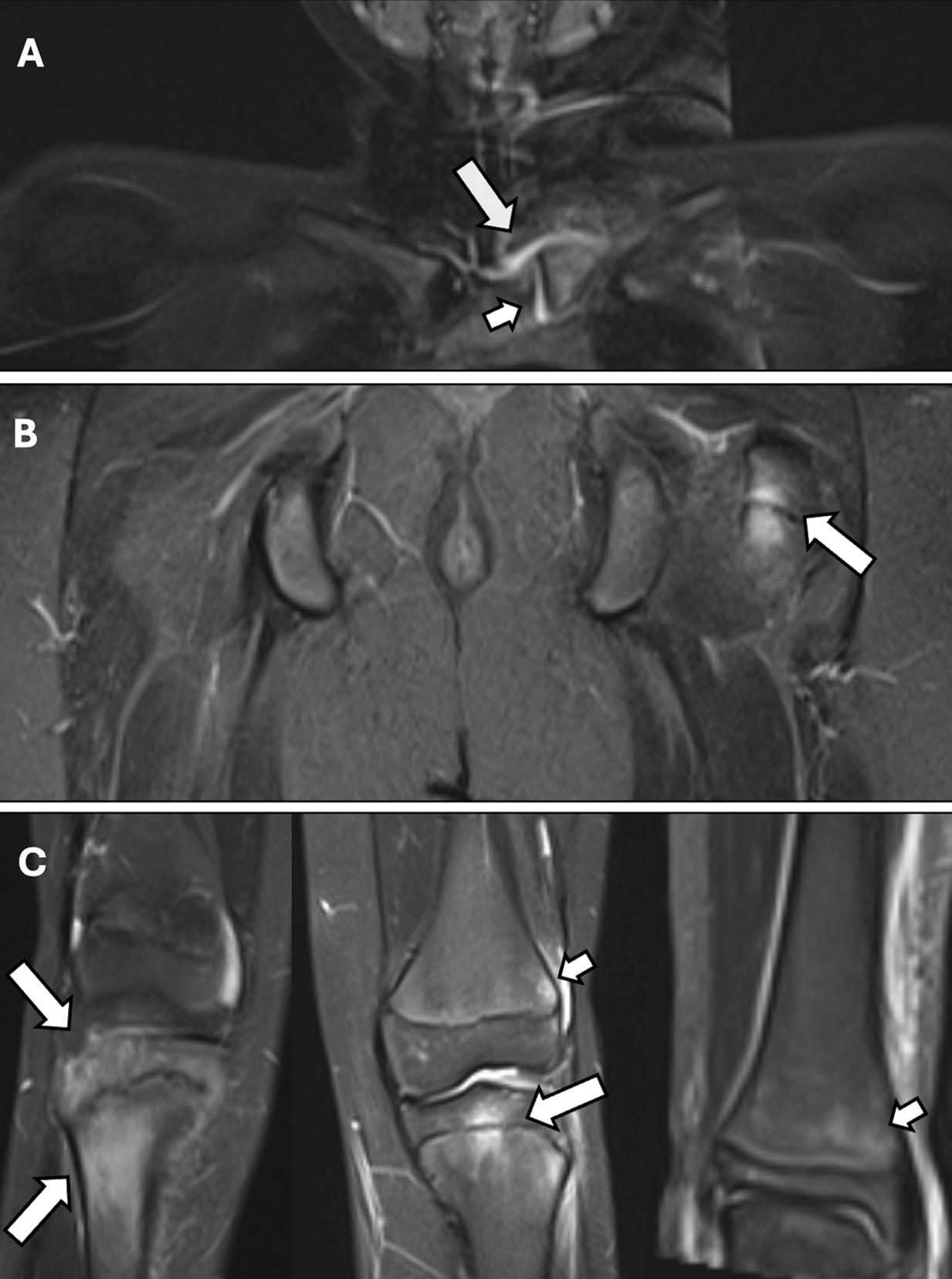
In addition to ongoing canakinumab, initial management included non-steroid anti-inflammatory drugs, followed by the introduction of sulfasalazine. However, approximately five months after starting sulfasalazine, he presented to the emergency department with severe abdominal pain. Laboratory tests showed elevated amylase and lipase levels, and abdominal computed tomography (CT) demonstrated findings consistent with acute pancreatitis. Drug-induced pancreatitis was considered the most likely etiology, and sulfasalazine was discontinued.
Given refractory systemic inflammation, persistent bone pain, and inadequate response to IL-1 inhibitors, canakinumab was discontinued and subcutaneous tocilizumab 162 mg/week (4 mg/kg/week) was initiated. The patient showed rapid improvement in systemic symptoms and bone pain, with normalization of inflammatory markers. Since approximately the third month of tocilizumab therapy, he has had no recurrence of bone pain. Over the past year, he experienced only a single mild flare characterized by fever and abdominal pain, and no evidence of subclinical inflammation has been detected, with acute-phase reactants consistently remaining within normal ranges. He has been maintained on weekly tocilizumab for 24 months and remains in sustained remission.
Written informed consent was obtained from the patient’s parents.
Discussion
The coexistence of MKD and CNO-like lesions highlights a significant overlap in autoinflammatory pathways, primarily mediated by the dysregulation of IL-1β, TNF-α, and IL-6.6,9,10 Rather than being a simple association of two separate entities, this case suggests a shared pathogenic theme of innate immune overactivation. While musculoskeletal manifestations such as arthralgia and transient arthritis are commonly observed during MKD flares, radiologically confirmed sterile osteitis resembling CNO has been only rarely reported in monogenic autoinflammatory diseases.9,10 This case illustrates an unusual clinical phenotype where both systemic inflammation and focal bone lesions responded completely to IL-6 inhibition.
Recent pediatric cohorts have broadened the phenotypic spectrum of MKD and highlighted its variable musculoskeletal involvement.1-5 Evaluating other autoinflammatory diseases associated with CNO-like bone involvement further strengthens this diagnostic framework. Given that our patient was initially suspected of having FMF, the relatively frequent association between FMF and CNO should be addressed.16 While this link is well-documented in FMF, MKD-associated CNO-like lesions remain extremely limited in the literature.6,10,16 Our patient expands this spectrum by demonstrating characteristic multifocal metaphyseal bone marrow edema on whole-body MRI. In the absence of a bone biopsy, the diagnostic value of MRI in CNO is paramount; the specific multifocal distribution and high-intensity signals on STIR sequences provided high diagnostic confidence, effectively supporting the diagnosis through non-invasive means.9,10,17
CNO is the most common autoinflammatory bone disease in childhood and is driven by dysregulated innate immunity.6,9 Evidence indicates that tocilizumab may be beneficial in refractory cases, supporting the pathogenic role of IL-6 in autoinflammatory bone inflammation.14,15 In parallel, IL-1 inhibitors are considered standard therapy for MKD; however, incomplete responses have been described, particularly in patients with multisystem involvement.2,11-13 Our patient demonstrated persistent inflammatory attacks and progressive bone pain despite sequential IL-1 blockade, indicating the need for alternative cytokine-directed therapy.
IL-6 contributes to osteoclast activation and bone marrow edema, providing a biologically plausible target in patients with MKD and concomitant bone involvement.8-10 Supportive evidence for IL-6 inhibition in MKD comes from small series showing reduced flare frequency under tocilizumab treatment.12,13 It should be noted that while many cited studies regarding IL-6 blockade describe primary CNO patients without MKD, we extrapolated from the established role of IL-6 in CNO to propose its potential effectiveness in MKD-associated bone lesions.14,15 The complete clinical and radiological remission achieved with tocilizumab in our patient suggests that IL-6 may play a key pathogenic role in both conditions, especially in phenotypes refractory to IL-1 inhibition.
This case demonstrates that radiologically confirmed CNO-like bone lesions can occur as a rare manifestation of mevalonate kinase deficiency. In pediatric patients presenting with autoinflammatory features and persistent bone pain, the use of whole-body MRI is a crucial, non-invasive diagnostic tool that can identify sterile osteitis without the need for bone biopsy. Furthermore, when standard IL-1 blockade fails to achieve full remission, IL-6 inhibition with tocilizumab should be considered as an effective therapeutic alternative for managing both systemic and musculoskeletal involvement in MKD. Further research into the shared cytokine pathways of these rare disorders may lead to more targeted and personalized treatment strategies.
Ethical approval
Written informed consent was obtained from the legal guardians of the patient. The signed consent forms are retained by the corresponding author.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- van der Burgh R, Ter Haar NM, Boes ML, Frenkel J. Mevalonate kinase deficiency, a metabolic autoinflammatory disease. Clin Immunol 2013; 147: 197-206. https://doi.org/10.1016/j.clim.2012.09.011
- Kılıç Könte E, Aslan E, Akay N, et al. Mevalonate kinase deficiency in a familial Mediterranean fever endemic region: a single-center experience. Turk J Pediatr 2025; 67: 483-492. https://doi.org/10.24953/turkjpediatr.2025.6015
- Lengvári L, Takács K, Lengyel A, et al. Mevalonate kinase deficiency: an updated clinical overview and revision of the SHARE recommendations. Front Immunol 2024; 15: 1466844. https://doi.org/10.3389/fimmu.2024.1466844
- Guan C, Wang W, Zhou Q, et al. Mevalonate kinase deficiency: genetic and clinical characteristics of a Chinese pediatric cohort. Pediatr Rheumatol Online J 2025; 23: 78. https://doi.org/10.1186/s12969-025-01131-1
- Gençpınar P, Makay BB, Gattorno M, Caroli F, Ünsal E. Mevalonate kinase deficiency (hyper IgD syndrome with periodic fever)--different faces with separate treatments: two cases and review of the literature. Turk J Pediatr 2012; 54: 641-644.
- Hedrich CM, Hofmann SR, Pablik J, Morbach H, Girschick HJ. Autoinflammatory bone disorders with special focus on chronic recurrent multifocal osteomyelitis (CRMO). Pediatr Rheumatol Online J 2013; 11: 47. https://doi.org/10.1186/1546-0096-11-47
- Zhao Y, Ferguson PJ. Chronic non-bacterial osteomyelitis and autoinflammatory bone diseases. Clin Immunol 2020; 216: 108458. https://doi.org/10.1016/j.clim.2020.108458
- Singhal S, Landes C, Shukla R, McCann LJ, Hedrich CM. Classification and management strategies for paediatric chronic nonbacterial osteomyelitis and chronic recurrent multifocal osteomyelitis. Expert Rev Clin Immunol 2023; 19: 1101-1116. https://doi.org/10.1080/1744666X.2023.2218088
- Maeda Y, Hiejima E, Izawa K, et al. The first nationwide epidemiological survey of chronic recurrent multifocal osteomyelitis in Japan. Mod Rheumatol 2025; 35: 1047-1056. https://doi.org/10.1093/mr/roaf045
- Haşlak F, Akay N, Gül Ü, et al. Autoinflammatory Bone Diseases. Balkan Med J 2025; 42: 5-13. https://doi.org/10.4274/balkanmedj.galenos.2024.2024-11-129
- Sánchez-Manubens J, Iglesias E, Anton J. Canakinumab for the treatment of hyperimmunoglobulin D syndrome. Expert Rev Clin Immunol 2019; 15: 215-220. https://doi.org/10.1080/1744666X.2019.1571410
- Rafiq NK, Lachmann H, Joensen F, Herlin T, Brogan PA. Tocilizumab for the Treatment of Mevalonate Kinase Deficiency. Case Rep Pediatr 2018; 2018: 3514645. https://doi.org/10.1155/2018/3514645
- Li C, Chen X, Tang X, Zeng H, Zhou J. Tocilizumab effectively reduces flares of hyperimmunoglobulin D syndrome in children: Three cases in China. Mol Genet Metab Rep 2024; 40: 101105. https://doi.org/10.1016/j.ymgmr.2024.101105
- Sato H, Wada Y, Hasegawa E, et al. Adult-onset Chronic Recurrent Multifocal Osteomyelitis with High Intensity of Muscles Detected by Magnetic Resonance Imaging, Successfully Controlled with Tocilizumab. Intern Med 2017; 56: 2353-2360. https://doi.org/10.2169/internalmedicine.8473-16
- Kaut S, Van den Wyngaert I, Christiaens D, et al. Chronic nonbacterial osteomyelitis in children: a multicentre Belgian cohort of 30 children. Pediatr Rheumatol Online J 2022; 20: 41. https://doi.org/10.1186/s12969-022-00698-3
- Çiçek SÖ, Şahin N, Karaman ZF, et al. The possible relationship between familial Mediterranean fever and chronic nonbacterial osteomyelitis: coincidence or coexistence? J Clin Rheumatol 2021; 27: e342-e348. https://doi.org/10.1097/RHU.0000000000001431
- Tunce E, Ulu K, Taşar S, Sözeri B. Utilization of whole-body magnetic resonance imaging in challenging diagnoses in pediatric rheumatology. Turk Arch Pediatr 2024; 59: 305-311. https://doi.org/10.5152/TurkArchPediatr.2024.23319
Copyright and license
Copyright © 2026 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









