
Graphical Abstract

Abstract
Background. Respiratory colonization with Pseudomonas aeruginosa is associated with increased morbidity and mortality in cystic fibrosis (CF) patients. This study aims to assess the clinical characteristics and associated factors of CF infants under two years of age with P. aeruginosa colonization in Türkiye.
Method. Of the 1637 patients registered in the Cystic Fibrosis Registry of Türkiye in 2019, 284 patients under two years of age were included in this retrospective cross-sectional study. Patients were classified into two groups: those with P. aeruginosa colonization (Group 1) and those without (Group 2). Cystic fibrosis transmembrane conductance regulator (CFTR) gene functions were categorized according to CFTR mutation functional class.
Results. Twenty-three patients (8.1%) were categorized as Group 1 and 262 participants (91.9%) were classified as Group 2. Infants with P. aeruginosa colonization (Group 1) were more likely to have minimal CFTR function compared with those without colonization (87% vs. 39.8%, p = 0.017). In addition, both Staphylococcus aureus colonization (47.8% vs. 7.3%, p < 0.001) and methicillin-resistant S. aureus positivity (17.4% vs. 6.1%, p = 0.042) were observed more commonly in Group 1. There were no statistical differences between the groups in terms of age at diagnosis, gender, mean z-scores of weight and height, newborn screening test positivity, sweat chloride test results, and pancreatic insufficiency (p > 0.05). Univariate logistic regression analysis did not identify significant associated factors for P. aeruginosa colonization.
Conclusions. Our findings suggest that minimal CFTR function and S. aureus colonization are associated with P. aeruginosa colonization in CF patients under two years of age. Further studies are needed to investigate associated factors for early P. aeruginosa colonization, eradication treatment effectiveness, and longitudinal outcomes of in CF patients under two years of age.
Keywords: Cystic fibrosis, Pseudomonas aeruginosa, colonization, children, registry
Introduction
Cystic fibrosis (CF) is the most common inherited disease in Caucasian populations caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which encodes the ion channel-associated CFTR protein.1 CFTR dysfunction leads to the colonization of an opportunistic Gram-negative pathogen, Pseudomonas aeruginosa.2 Chronic infection with P. aeruginosa is one of the leading causes of morbidity and mortality in CF patients by contributing to progressive decline in pulmonary function.2 With new modulator therapies, advances in supportive care, and enhanced treatment of respiratory infections and other complications, the predicted life expectancy of CF patients has increased to almost 50 years.3 Therefore, recognition and effective treatment of initial P. aeruginosa colonization is essential in maintaining lung health and long-life expectancy in children with CF.4
Longitudinal studies in children with CF assessed with bronchoscopy and oropharyngeal cultures have demonstrated a high prevalence of P. aeruginosa in the first 2 years of life.5-8 Moreover, although chronic P. aeruginosa colonization is not commonly expected during the first years of life, very early acquisition in infancy represents a critical and vulnerable period, as initial airway colonization during this stage may have a disproportionate impact on subsequent disease severity and long-term outcomes, including mortality in young children with CF.7,9,10 However, the specific risk factors and clinical implications of P. aeruginosa colonization in infants remain inadequately understood, particularly in populations with genetic and environmental diversity. The fact that the prevalence of P. aeruginosa is not decreasing in our country, unlike in the United Kingdom, the United States, and European countries, makes the investigation of infant CF patients with P. aeruginosa colonization more important.11-14
Based on our hypothesis that very early P. aeruginosa colonization during the first years of life represents a distinct and sensitive clinical entity, we specifically focused on infants under 2 years of age. In this retrospective cross-sectional study, we aimed to assess the clinical features of CF patients with P. aeruginosa colonization under 2 years of age, in order to evaluate the associated factors for P. aeruginosa colonization.
Methods
Study design
We conducted a retrospective cross-sectional study on children with CF aged under 2 years of age who had available data on P. aeruginosa colonization status in the Cystic Fibrosis Registry of Türkiye (CFRT) for 2019. Patients aged <2 years were divided into two groups: those with P. aeruginosa colonization (Group 1) and those without P. aeruginosa colonisation (Group 2). All analyses were compared with these two groups.
Data input to the registry was approved by the local ethics committee, and all patients and/or their parents signed written consent for the data entry. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee (Hacettepe University Ethics Board, date: 12 April 2007, reference number: HEK 07/16-21 and date: 5 June 2018, reference number: GO 18/473-31) and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Diagnosis of P. aeruginosa colonization in cystic fibrosis
Cystic fibrosis diagnosis was established according to the European Cystic Fibrosis Society (ECFS) guideline.15 Chronic P. aeruginosa colonization was defined according to “modified Leeds criteria” as applied in the ECFSPR guideline: > 50% of the samples (sputum/other) collected over a 12-month period should be positive; at least 4 samples collected.15,16 Samples were obtained at each outpatient clinic visit or during hospital stays using cough swabs or oropharyngeal swabs, as the study population was unable to expectorate sputum, as reported in the literature.7,17 Infants are recommended to attend an outpatient clinic visits within the first 3 months of life after the diagnosis, then every 3-6 months until 2 years of age.18
Data variables
Age at the study period, age at diagnosis, gender, weight, height, z-scores of weight and height, history of meconium ileus, newborn screening test results, sweat chloride test results, medications, and results of CFTR genotype analysis were noted. As recommended by the CDC, Z-scores of weight and height measurements were assessed by using the World Health Organization growth charts for children < 24 months of age.19,20 CF patients with classic symptoms and signs of exocrine pancreatic insufficiency who also have fecal elastase values <200 μg/g are said to have pancreatic insufficiency.15
CFTR functions were categorized according to CFTR mutation functional class: minimal function (presence of only class I, II, or III mutations) and residual function (at least one class IV or V mutation).21-23 Minimal function mutations are severe and commonly associated with advanced lung disease and pancreatic insufficiency, whereas residual function mutations are milder and linked to less severe clinical phenotypes.
The presence of microorganisms in respiratory cultures recorded during the 2019 registry year, such as Staphylococcus aureus, methicillin-resistant S. aureus (MRSA), and Haemophilus influenzae, Stenotrophomonas maltophilia, and Achromobacter species were noted. The colonization status of P. aeruginosa, S. aureus, and the Burkholderia cepacia complex were recorded. The data regarding associated complications, including pseudo-Bartter syndrome (PBS), CF-related liver disease, gastroesophageal reflux, sinusitis, pneumothorax, and hemoptysis, were also documented.
Statistical analysis
Statistical Package for the Social Sciences (SPSS for Windows Version 20) was used for statistical analyses. In the descriptive statistics section, categorical variables are presented with numbers, percentages, and continuous variables with mean ± standard deviation or median and interquartile range (IQR, Q1-Q3). The distribution of normality in groups was evaluated by Kolmogorov-Smirnov and Shapiro-Wilk tests. A comparison of the median values of two independent groups was measured by the Mann-Whitney U test. The percentage distribution of categorical data between groups was measured using the χ2 test. Values of p < 0.05 were considered statistically significant. Because of the limited number of outcome events, logistic regression analyses were restricted to univariate models.
Results
A total of 1637 patients were registered in the Cystic Fibrosis Registry of Türkiye in 2019. Among these, 284 (17.3%) patients under 2 years of age were included in this retrospective cross-sectional study.
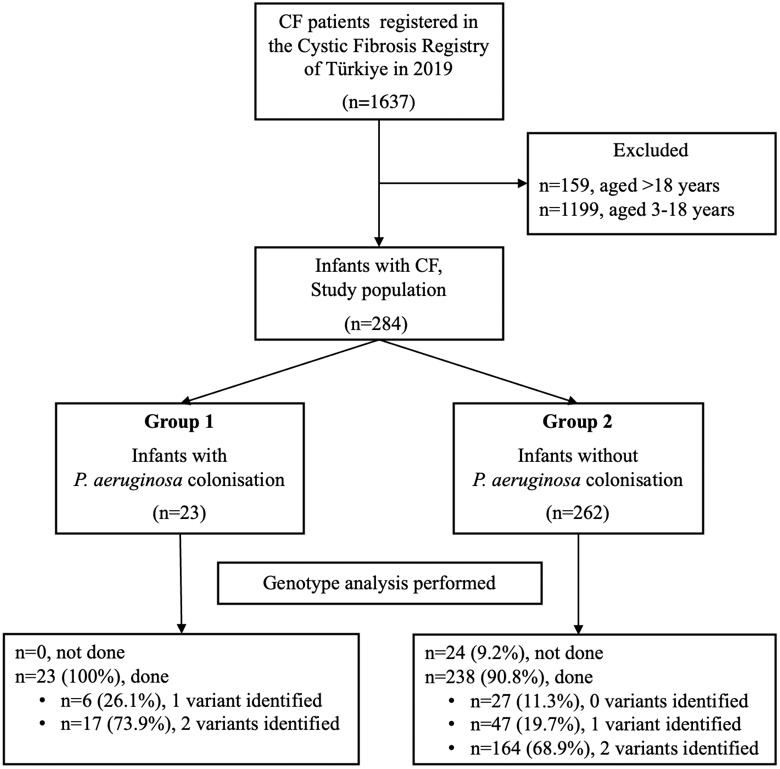
In the study population (n = 284), 23 participants (8.1%) were assigned to group 1, while 262 participants (91.9%) were assigned to group 2. The eligibility assessment of patients included in the study is shown in Fig. 1.
Demographic and clinical characteristics
Demographic and clinical characteristics of the study population according to P. aeruginosa colonization status are shown in Table I. Since newborn screening for CF was introduced in Türkiye on January 1st, 2015, all patients had undergone newborn screening. Among those screened, 17 patients (73.9%) in Group 1 and 201 patients (77.0%) in Group 2 had a positive newborn screening result. There were no statistical differences between the groups in terms of age, age at diagnosis, gender, mean z-scores of weight and height, newborn screening test positivity, sweat chloride test results, and pancreatic insufficiency (p > 0.05).
|
*Age at the time of data entry for the 2019 registry year. aGroup comparisons were done using Mann‐Whitney U test. bGroup comparisons were done using Student’s t test CFTR: cystic fibrosis transmembrane conductance regulator, IQR: interquartile range, SD: standard deviation. |
|||
| Table I. Comparison of demographic and clinical features of patients with cystic fibrosis under 2 years of age according to P. aeruginosa colonization. | |||
| Variables |
P. aeruginosa colonization (n = 23) |
P. aeruginosa colonization (n = 262) |
|
| Age at diagnosis, months, median (IQR) |
|
|
|
| Age*, months, median (IQR) |
|
|
|
| Gender, male, n (%) |
|
|
|
| Weight, kg, mean ± SD |
|
|
|
| Height, cm, mean ± SD |
|
|
|
| z-score for weight for age, mean ± SD |
|
|
|
| z-score for height, mean ± SD |
|
|
|
| Diagnosis by newborn screening, n (%) |
|
|
|
| History of meconium ileus, n (%) |
|
|
|
| Sweat chloride test, mmol/L, mean ± SD |
|
|
|
| Pancreatic insufficiency, n (%) |
|
|
|
| CFTR genotype analysis, done, n (%) |
|
|
|
| Homozygous F508del genotype, n (%) |
|
|
|
| Heterozygous F508del genotype, n (%) |
|
|
|
| CFTR classification, n (%) |
|
|
|
| Minimal function |
|
|
|
| Residual function |
|
|
|
| Unclassified |
|
|
|
CFTR genotype analysis
Genotype analysis was performed on all patients in Group 1 (Fig. 1). F508del homozygous and heterozygous mutations were present in one patient each. According to the CFTR functional classification, 20 (87%) patients had minimal CFTR function, and 1 (4.8%) had residual CFTR function.
In Group 2, at least one variant was detected in 201 of the 238 patients who were genotyped (Fig. 1). Nineteen patients (7.3%) were homozygous for the F508del mutation, and 38 (14.5%) were heterozygous for F508del. According to the CFTR functional classification, 84 patients (39.8%) had minimal CFTR function, and 35 (16.6%) had residual CFTR function. There was no statistical difference between the two groups regarding F508del homozygous and heterozygous mutations (p > 0.05). Patients in group 1 had a higher prevalence of minimal CFTR function than those in group 2 (p = 0.017).
Microbiological features
The prevalence of S. aureus (58.3% vs. 23.9%, p = 0.007) and MRSA (17.4% vs. 6.1%, p = 0.042) on recent respiratory culture and S. aureus colonization (47.8% vs 7.3%, p < 0.001) were significantly higher in Group 1, compared with Group 2 (Table II).
|
*Data given as number (percentage), except for annual IV antibiotic days due to PEx, presented as median (interquartile range). **Presence of microorganisms in respiratory cultures during the 2019 registry year aGroup comparisons were done using Mann‐Whitney U test. CF: cystic fibrosis, IV: intravenous, MRSA: methicillin-resistant Staphylococcus aureus, PEx: pulmonary exacerbations |
|||
| Table II. Treatments, microbiological results, and accompanying complications of the study cohort of cystic fibrosis patients under 2 years of age. | |||
|
P. aeruginosa colonization |
P. aeruginosa colonization |
|
|
| Treatment | |||
| DNase |
|
|
|
| Vitamin |
|
|
|
| Pancreatic enzymes |
|
|
|
| Inhaled hypertonic saline |
|
|
|
| Inhaled antibiotics |
|
|
|
| Oxygen therapy |
|
|
|
| Annual IV antibiotics days due to PEx, median (IQR)* |
|
|
|
| Microbiological findings** | |||
| Haemophilus influenzae |
|
|
|
| Staphylococcus aureus |
|
|
|
| MRSA |
|
|
|
| Stenotrophomonas maltophilia |
|
|
|
| Achromobacter |
|
|
|
| Chronic colonization | |||
| Staphylococcus aureus |
|
|
|
| Burkholderia cepacia |
|
|
|
| Complications | |||
| Pseudo-Bartter syndrome |
|
|
|
| CF-related liver disease |
|
|
|
| Gastroesophageal reflux |
|
|
|
| Sinusitis |
|
|
|
Both P. aeruginosa and S. aureus colonization were present in five patients. In three cases, S. aureus colonization preceded P. aeruginosa colonization.
CF-related medical treatments and CF-related complications
Patients in Group 1 had statistically longer duration of antibiotic therapy for pulmonary exacerbations (15.1 vs. 4.5, p < 0.001), more oxygen therapy support (8.7% vs. 0.8%, p = 0.034), higher prevalence of DNase (100% vs. 82.7%, p = 0.003), inhaled hypertonic saline (13% vs. 3.8%, p = 0.042), and inhaled antibiotic treatments (26.1% vs. 1.9%, p < 0.001) than patients in Group 2. A comparison of the treatments, microbiological results, and accompanying complications of the groups is given in Table II.
There were no significant differences between the groups in terms of accompanying complications, including PBS, CF-related liver disease (elevated transaminases without cirrhosis), gastroesophageal reflux, and sinusitis (p > 0.05). No other complications were recorded in any patients (Table II). None of the patients in the study cohort were receiving CFTR modulator therapy during the study period.
In the univariate logistic regression analyses, no relationship was found between gender, age at diagnosis, newborn screening test positivity, sweat chloride test results, and pancreatic insufficiency on the probability of P. aeruginosa colonization.
Discussion
We described the characteristics of infants under 2 years of age with P. aeruginosa colonization in our country. The prevalence of P. aeruginosa colonization was found to be 8.1% under 2 years of age. S. aureus infection and colonization were significantly higher in patients with P. aeruginosa colonization. We demonstrated P. aeruginosa colonization was more frequent in infants with minimal CFTR function compared to those with residual CFTR function.
Age-specific data on P. aeruginosa colonization in early childhood are limited and inconsistently reported in annual registry summaries.13,15,24-27 For example, the prevalence of P. aeruginosa colonization has been reported as 2.1% in children under 3 years of age in the United Kingdom CF Registry and 6% in children under 4 years of age in the French CF Registry.13,27 In contrast, the overall prevalence of P. aeruginosa colonization across all pediatric age groups has been reported as approximately 3.3% in the UK and 20% in France. In this context, our study provides age-specific data for infants under 2 years of age and demonstrates a prevalence of chronic P. aeruginosa colonization of 8.1% in this early and vulnerable period, compared with an overall colonization rate of 20% reported in the national registry.24
The age at initial P. aeruginosa acquisition is likely influenced by a complex interaction of bacterial, host-related (especially genetics), and environmental factors.4,28-30 Pulmonary microbial diversity—defined as the richness and relative abundance of different microorganisms within the airway—is highest in early childhood and has been shown to influence disease progression and susceptibility to early P. aeruginosa colonization in patients with CF.28,31 Moreover, studies have shown that increased airway inflammation, even in the absence of overt infection, may lead to early tissue damage and create a permissive environment for subsequent P. aeruginosa acquisition even in early infancy.4,32 These mechanisms may partly explain why early-life colonization occurs in a particularly vulnerable period of lung development.
In infants who do not expectorate sputum, respiratory surveillance commonly relies on oropharyngeal or cough swabs to identify P. aeruginosa.15,33 Although concerns remain about their diagnostic accuracy, a randomized controlled study demonstrated comparable clinical outcomes between bronchoalveolar lavage–guided and oropharyngeal culture–guided treatment strategies in young children with CF.34 Manos et al. showed that persistent P. aeruginosa strains in infants varied independently from isolation sites, such as upper or lower airways, and prior exposure of the airway to P. aeruginosa.35 Taken together, these findings suggest that although the specific methods used for respiratory sample collection were not available in our study, the interpretation of our results remains reliable and consistent with existing evidence.
Patient characteristics such as lower socio-economic status, female gender, CFTR genotype, and microbial diversity in the lungs have been associated or identified as risk factors for the initial acquisition of P. aeruginosa. 4,10,17,30,36,37 Rosenfeld et al. evaluated risk factors for initial P. aeruginosa acquisition in order to inform prevention strategies and identify high-risk populations.30 They concluded that none of the modifiable risk factors evaluated, including cigarette smoke, hot tub use, breastfeeding, and newborn screening positivity, was associated with age at P. aeruginosa acquisition. Rosenfeld and other studies have demonstrated that minimal CFTR function is associated with earlier P. aeruginosa acquisition compared to those with residual CFTR function.23,30 Most of these studies have been conducted in the US, where the frequency of the F508del mutation is over 80%. However, due to the mutation diversity in our country, the frequency of the F508del mutation, the most common variant, is only 25%.24 In the only study conducted in our country on CFTR function classification, no association was found between minimal and residual CFTR function classification and FEV1 decline.38 However, we found the frequency of P. aeruginosa colonization was higher in those with minimal CFTR function compared to those with residual CFTR function under the age of 2 years. This causality analysis is beyond the scope of our study; however, the potential correlation between CFTR function classification in CF patients and disease severity at follow-up, including the risk of P. aeruginosa colonization, represents a significant research question for future investigations in our country.
The prevalence of S. aureus and MRSA isolation on recent respiratory culture positivity and S. aureus colonization were significantly higher in patients with P. aeruginosa colonization under 2 years of age. The relationship between P. aeruginosa and S. aureus in patients with CF is controversial in the literature. While Maselli et al. showed that S. aureus is a risk factor for initial P. aeruginosa acquisition, another study demonstrated S. aureus infection is significantly lower in the chronic P. aeruginosa group.16,17 In our study, although higher rates of S. aureus and MRSA were observed in infants with P. aeruginosa colonization, the limited sample size precluded robust statistical inference regarding the nature of this association. Therefore, larger studies are required to better clarify the relationship between S. aureus and early P. aeruginosa colonization.
CF registries are a valuable resource for research because they provide access to large populations; however, they also have inherent limitations due to the restricted scope of available data. By focusing on a single time point, we were unable to evaluate the long-term progression of P. aeruginosa colonization and its impact on pulmonary function over time. An important limitation of the present study is the lack of detailed data on eradication therapies and treatment outcomes, which precluded evaluation of the effectiveness of different therapeutic approaches in preventing chronic P. aeruginosa colonization. Similarly, environmental factors such as air quality and climate were not assessed, which may play a role in colonization variability across regions and centers. Future studies incorporating genetic analysis, environmental factors, and documenting treatment regimens could yield more comprehensive risk assessments. Another limitation is that our study preceded the widespread availability of CFTR modulators in Türkiye. The absence of patients receiving these therapies in our cohort may limit the generalizability of our findings to current cystic fibrosis management, as these treatments may potentially alter airway microbiology. The relatively small number of infants with P. aeruginosa colonization reduced statistical power and limited the ability to perform reliable multivariable analyses, increasing the risk of type II error. Therefore, logistic regression was restricted to a small number of clinically relevant, non-treatment variables, and the absence of significant independent predictors should be interpreted with caution. Although the use of standardized Leeds criteria for colonization diagnosis enhances the study’s reliability, longitudinal follow-up studies are essential to evaluate the persistence of colonization, the effectiveness of interventions, and to clarify causal relationships and risk factors.
In conclusion, this study highlights the clinical characteristics and associated factors associated with P. aeruginosa colonization in infants with CF in Türkiye. The higher prevalence of colonization compared to other countries underscores the need for targeted preventive strategies. Our findings suggest that minimal CFTR function may contribute to an increased risk of colonization, and the association with S. aureus colonization further emphasizes the complexity of early respiratory infections in CF patients. However, the lack of data on eradication therapies and treatment outcomes limits our ability to assess intervention effectiveness. Future longitudinal studies incorporating genetic, environmental, and microbiome analyses are essential to better understand the dynamics of P. aeruginosa acquisition and to improve early intervention strategies in CF infants.
Acknowledgements
The authors are grateful to the Cystic Fibrosis Registry of Türkiye for providing access to the data of patients and thank the individual center representatives for allowing the use of data.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee (Hacettepe University Ethics Board, date: 12 April 2007, reference number: HEK 07/16-21 and date: 5 June 2018, reference number: GO 18/473-31) and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Ratjen F, Bell SC, Rowe SM, Goss CH, Quittner AL, Bush A. Cystic fibrosis. Nat Rev Dis Primers 2015; 1: 15010. https://doi.org/10.1038/nrdp.2015.10
- Murray TS, Egan M, Kazmierczak BI. Pseudomonas aeruginosa chronic colonization in cystic fibrosis patients. Curr Opin Pediatr 2007; 19: 83-88. https://doi.org/10.1097/MOP.0b013e3280123a5d
- McBennett KA, Davis PB, Konstan MW. Increasing life expectancy in cystic fibrosis: advances and challenges. Pediatr Pulmonol 2022; 57(Suppl 1): S5-S12. https://doi.org/10.1002/ppul.25733
- Jackson L, Waters V. Factors influencing the acquisition and eradication of early Pseudomonas aeruginosa infection in cystic fibrosis. J Cyst Fibros 2021; 20: 8-16. https://doi.org/10.1016/j.jcf.2020.10.008
- Burns JL, Gibson RL, McNamara S, et al. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J Infect Dis 2001; 183: 444-452. https://doi.org/10.1086/318075
- Armstrong DS, Grimwood K, Carlin JB, Carzino R, Olinsky A, Phelan PD. Bronchoalveolar lavage or oropharyngeal cultures to identify lower respiratory pathogens in infants with cystic fibrosis. Pediatr Pulmonol 1996; 21: 267-275. https://doi.org/10.1002/(SICI)1099-0496(199605)21:5<267::AID-PPUL1>3.0.CO;2-K
- Hudson VL, Wielinski CL, Regelmann WE. Prognostic implications of initial oropharyngeal bacterial flora in patients with cystic fibrosis diagnosed before the age of two years. J Pediatr 1993; 122: 854-860. https://doi.org/10.1016/s0022-3476(09)90007-5
- Savant AP, McColley SA. Cystic fibrosis year in review 2019: section 2 pulmonary disease and infections. Pediatr Pulmonol 2023; 58: 672-682. https://doi.org/10.1002/ppul.25091
- Nixon GM, Armstrong DS, Carzino R, et al. Clinical outcome after early Pseudomonas aeruginosa infection in cystic fibrosis. J Pediatr 2001; 138: 699-704. https://doi.org/10.1067/mpd.2001.112897
- Demko CA, Byard PJ, Davis PB. Gender differences in cystic fibrosis: Pseudomonas aeruginosa infection. J Clin Epidemiol 1995; 48: 1041-1049. https://doi.org/10.1016/0895-4356(94)00230-n
- European Cystic Fibrosis Society Patient Registry 2022 Annual Data Report. 2022. Available at: https://www.ecfs.eu/sites/default/files/Annual%20Report_2022_ECFSPR_20240603.pdf (Accessed on Jan 1, 2025).
- Ulusal kistik fibrozis kayıt sistemi 2023 yılı verileri. 2023. Available at: https://www.kistikfibrozisturkiye.org/wp-content/uploads/2024/11/2023-UKKS-2.pdf (Accessed on Jan 8, 2025).
- UK Cystic Fibrosis Registry Annual Data Report 2022. 2022. Available at: https://www.cysticfibrosis.org.uk/sites/default/files/2023-10/CFT_2022_Annual_Data_Report_FINAL_v8.pdf (Accessed on Jan 1, 2025).
- Cystic Fibrosis Foundation Patient Registry Annual Data Report 2023. Available at: https://www.cff.org/sites/default/files/2024-06/2023-Cystic-Fibrosis-Foundation-Patient-Registry-Highlights-Handout.pdf (Accessed on Jan 1, 2025).
- ECFS Patient Registry. Available at: https://www.ecfs.eu/ecfspr/ (Accessed on Feb 20, 2020).
- Proesmans M, Balinska-Miskiewicz W, Dupont L, et al. Evaluating the “Leeds criteria” for Pseudomonas aeruginosa infection in a cystic fibrosis centre. Eur Respir J 2006; 27: 937-943. https://doi.org/10.1183/09031936.06.00100805
- Maselli JH, Sontag MK, Norris JM, MacKenzie T, Wagener JS, Accurso FJ. Risk factors for initial acquisition of Pseudomonas aeruginosa in children with cystic fibrosis identified by newborn screening. Pediatr Pulmonol 2003; 35: 257-262. https://doi.org/10.1002/ppul.10230
- T.C. Sağlık Bakanlığı Türk Halk Sağlığı Kurumu. Kistik fibrozis yenidoğan tarama testi ile tanı alan hastaları izleme rehberi. 2017. Available at: https://hsgm.saglik.gov.tr/depo/birimler/cocuk_ergen_db/dokumanlar/yayinlar/Kitaplar/KF_Rehberi.pdf (Accessed on Mar 1, 2020).
- A SAS Program for the 2000 CDC Growth Charts (ages 0 to <20 years). Available at: https://www.cdc.gov/growth-chart-training/hcp/computer-programs/sas.html (Accessed on Feb 25, 2022).
- World Health Organization (WHO). WHO child growth standards: length/height-for-age, weight-for-age, weight-for-length, weight-for-height and body mass index-for-age: methods and development. Available at: https://apps.who.int/iris/bitstream/handle/10665/43413/924154693X_eng.pdf (Accessed on Mar 12, 2021).
- Green DM, McDougal KE, Blackman SM, et al. Mutations that permit residual CFTR function delay acquisition of multiple respiratory pathogens in CF patients. Respir Res 2010; 11: 140. https://doi.org/10.1186/1465-9921-11-140
- McKone EF, Goss CH, Aitken ML. CFTR genotype as a predictor of prognosis in cystic fibrosis. Chest 2006; 130: 1441-1447. https://doi.org/10.1378/chest.130.5.1441
- McKone EF, Emerson SS, Edwards KL, Aitken ML. Effect of genotype on phenotype and mortality in cystic fibrosis: a retrospective cohort study. Lancet 2003; 361: 1671-1676. https://doi.org/10.1016/S0140-6736(03)13368-5
- Ulusal kistik fibrozis kayıt sistemi 2019 yıllık verileri. 2019. Available at: https://www.kistikfibrozisturkiye.org/hasta-kayit-sistemi/#yillik-raporlar (Accessed on Feb 22, 2022).
- French Cystic Fibrosis Registry Annual Data Report 2019. 2021. Available at: https://www.vaincrelamuco.org/sites/default/files/french_cf_registry_2019_annual_report.pdf (Accessed on Jul 1, 2021).
- Cystic Fibrosis Foundation Patient Registry 2019 Annual Data Report. 2020. Available at: https://www.cff.org/sites/default/files/2021-10/2019-Patient-Registry-Annual-Data-Report.pdf (Accessed on Dec 1, 2020).
- French Cystic Fibrosis Registry Annual Data Report 2021. Available at: https://www.vaincrelamuco.org/sites/default/files/french_cf_registry_annual_data_report_2021.pdf (Accessed on Jan 1, 2023).
- Martiniano SL, Elbert AA, Farrell PM, et al. Outcomes of infants born during the first 9 years of CF newborn screening in the United States: a retrospective Cystic Fibrosis Foundation Patient Registry cohort study. Pediatr Pulmonol 2021; 56: 3758-3767. https://doi.org/10.1002/ppul.25658
- Pillarisetti N, Williamson E, Linnane B, et al. Infection, inflammation, and lung function decline in infants with cystic fibrosis. Am J Respir Crit Care Med 2011; 184: 75-81. https://doi.org/10.1164/rccm.201011-1892OC
- Rosenfeld M, Emerson J, McNamara S, et al. Risk factors for age at initial Pseudomonas acquisition in the cystic fibrosis epic observational cohort. J Cyst Fibros 2012; 11: 446-453. https://doi.org/10.1016/j.jcf.2012.04.003
- Frayman KB, Armstrong DS, Carzino R, et al. The lower airway microbiota in early cystic fibrosis lung disease: a longitudinal analysis. Thorax 2017; 72: 1104-1112. https://doi.org/10.1136/thoraxjnl-2016-209279
- Tirouvanziam R, de Bentzmann S, Hubeau C, et al. Inflammation and infection in naive human cystic fibrosis airway grafts. Am J Respir Cell Mol Biol 2000; 23: 121-127. https://doi.org/10.1165/ajrcmb.23.2.4214
- Guy’s and St Thomas’ NHS Foundation Trust. Clinical Guidelines: Care of Children Cystic Fibrosis Royal Brompton Hospital. Available at: https://www.rbht.nhs.uk/childrencf (Accessed on Jan 1, 2025).
- Wainwright CE, Vidmar S, Armstrong DS, et al. Effect of bronchoalveolar lavage-directed therapy on Pseudomonas aeruginosa infection and structural lung injury in children with cystic fibrosis: a randomized trial. JAMA 2011; 306: 163-171. https://doi.org/10.1001/jama.2011.954
- Manos J, Hu H, Rose BR, et al. Virulence factor expression patterns in Pseudomonas aeruginosa strains from infants with cystic fibrosis. Eur J Clin Microbiol Infect Dis 2013; 32: 1583-1592. https://doi.org/10.1007/s10096-013-1916-7
- Kosorok MR, Jalaluddin M, Farrell PM, et al. Comprehensive analysis of risk factors for acquisition of Pseudomonas aeruginosa in young children with cystic fibrosis. Pediatr Pulmonol 1998; 26: 81-88. https://doi.org/10.1002/(sici)1099-0496(199808)26:2<81::aid-ppul2>3.0.co;2-k
- Wang SS, FitzSimmons SC, O’Leary LA, Rock MJ, Gwinn ML, Khoury MJ. Early diagnosis of cystic fibrosis in the newborn period and risk of Pseudomonas aeruginosa acquisition in the first 10 years of life: A registry-based longitudinal study. Pediatrics 2001; 107: 274-279. https://doi.org/10.1542/peds.107.2.274
- Emiralioğlu N, Çakır B, Sertçelik A, et al. Factors associated with pulmonary function decline of patients in the cystic fibrosis registry of Turkey: a retrospective cohort study. Pediatr Pulmonol 2024; 59: 2956-2966. https://doi.org/10.1002/ppul.27165
Copyright and license
Copyright © 2026 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









