
Graphical Abstract
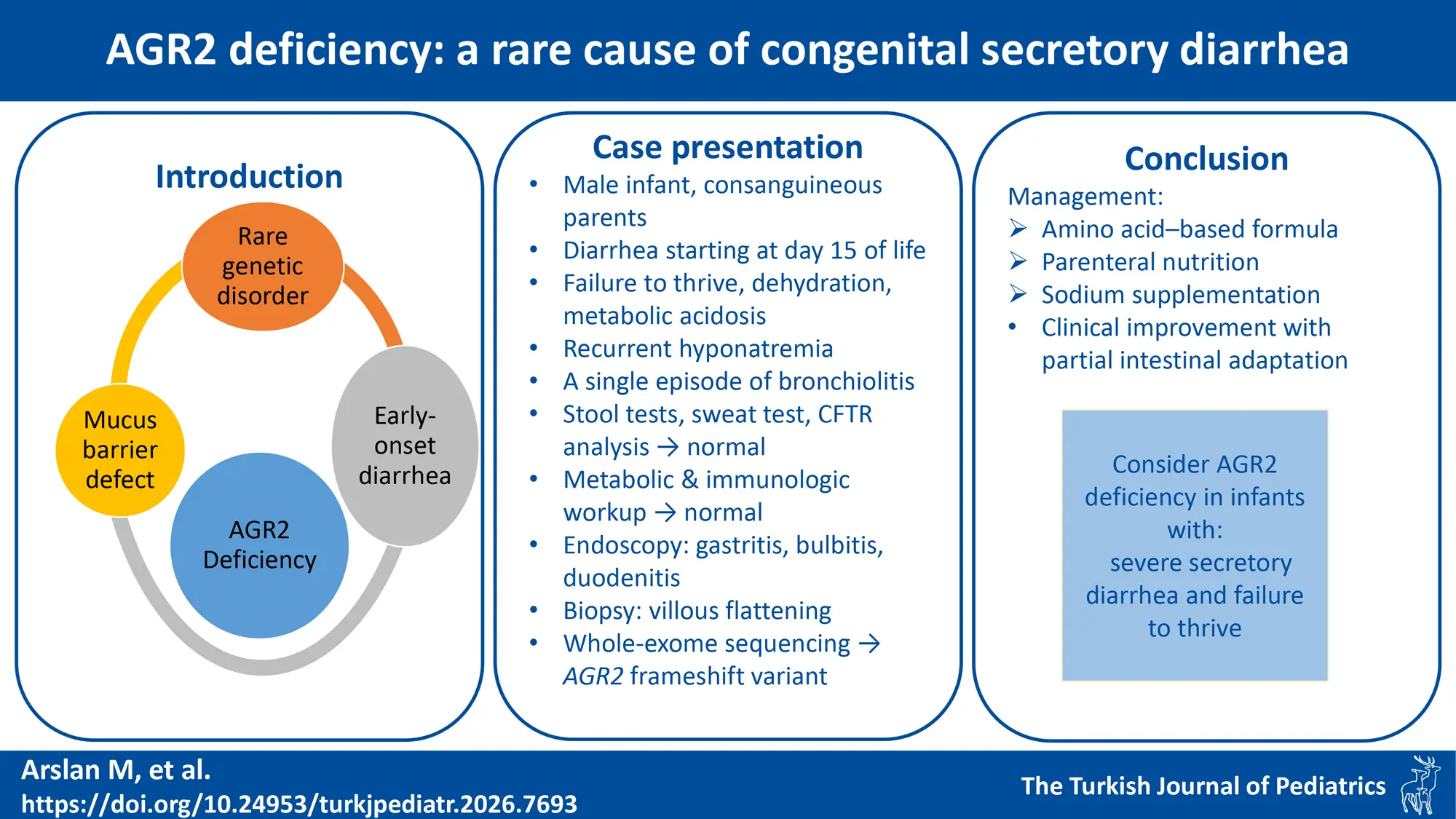
Abstract
Background. Congenital diarrheal disorders and enteropathies (CODE) are rare genetic conditions presenting with severe diarrhea and growth failure beginning in infancy. Biallelic variants in the anterior gradient 2 (AGR2) gene have recently been associated with a cystic fibrosis–like disorder characterized by multisystem involvement, including gastrointestinal and respiratory manifestations.
Case Presentation. We report a male infant born to consanguineous parents, who presented with severe congenital secretory diarrhea starting in the neonatal period, failure to thrive, dehydration, and metabolic acidosis. The diarrhea was refractory to bowel rest, suggesting a secretory mechanism. Stool studies, fecal elastase, sweat chloride testing, and CFTR gene analysis were normal. Endoscopic evaluation revealed antral gastritis, bulbitis, and duodenitis, with duodenal biopsies showing villous flattening. The patient developed recurrent hyponatremia requiring prolonged oral sodium supplementation and experienced a single episode of bronchiolitis, which may represent either a possible early respiratory manifestation or a coincidental common infantile infection. Whole exome sequencing identified a homozygous frameshift variant in the AGR2 gene (c.247_250del; p.His83ValfsTer4), and parental testing confirmed autosomal recessive inheritance.
Conclusion. This case expands the clinical spectrum of AGR2-related disease by highlighting a predominantly gastrointestinal presentation with severe congenital secretory diarrhea, failure to thrive, electrolyte imbalance, and a possible early respiratory manifestation. Given the rarity of this condition, AGR2 deficiency may be considered in selected infants presenting with severe congenital diarrhea and failure to thrive, particularly in the setting of consanguinity, even in the absence of definite pulmonary involvement.
Keywords: AGR2, congenital diarrhea, failure to thrive, infant, mucus barrier
Introduction
Congenital diarrheal disorders and enteropathies (CODE) are a group of rare diseases that primarily affect intestinal epithelial cell function, leading to diarrhea and impaired growth beginning in infancy. Affected patients often require lifelong fluid and nutritional support.1 As most cases of CODE are associated with single-gene defects, their prevalence is increased in populations in which consanguineous marriages are common. Next-generation sequencing has been used increasingly in recent years as a powerful tool to identify known and novel pathogenic variants causing congenital diarrhea.1,2
AGR2, encoded by the anterior gradient 2 (AGR2; MIM *606358) gene, is an endoplasmic reticulum resident-protein disulfide isomerase that catalyzes disulfide bond formation between cysteine residues during protein folding.3 AGR2 is highly expressed in mucus-secreting tissues, including the lungs and gastrointestinal tract. Secreted mucins, the main components of mucus, are high–molecular-weight glycoproteins containing numerous cysteine residues that facilitate proper folding and multimerization through disulfide bond formation, and AGR2 is required for the correct processing of gel-forming mucins.3,4 Biallelic AGR2 variants have recently been identified as the cause of a cystic fibrosis–like disorder characterized by recurrent respiratory infections and failure to thrive, with or without diarrhea (RIFTD; MIM #620233).3,5 To date, only a very limited number of patients with AGR2-related disease have been reported, highlighting the rarity of this condition and the need for further clinical descriptions.
In this case report, we report an infant from consanguineous parents, who presented with early-onset congenital diarrhea and failure to thrive, in whom exome sequencing identified a homozygous frameshift variant in the AGR2 gene.
Case Presentation
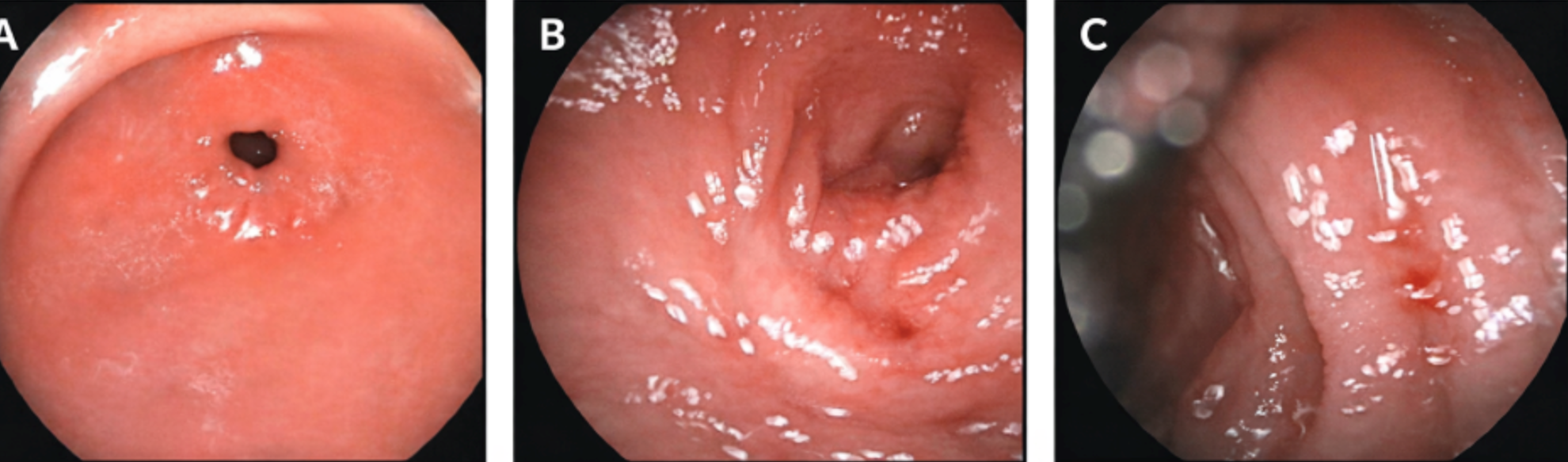
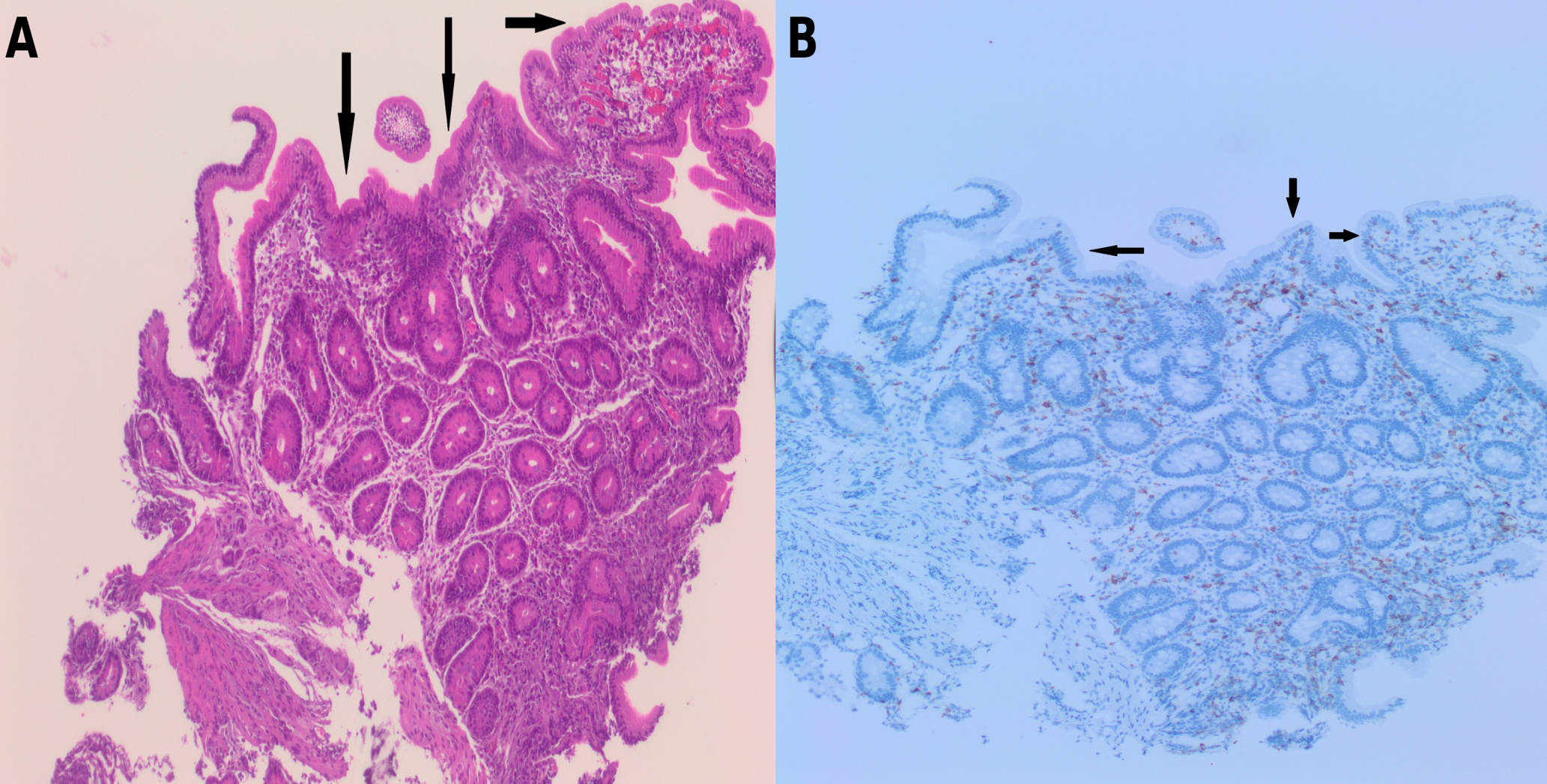
The patient was a male infant born at 38 weeks of gestation with a birth weight of 2850 g to consanguineous parents (first-degree cousins) of Syrian origin. There was no family history of chronic diarrhea, recurrent infections, or other known genetic disorders. He was referred to our hospital at 2 months of age with persistent diarrhea and failure to thrive that had started on postnatal day 15. On admission, his weight was 3100 g. Physical examination revealed dehydration, abdominal distension, and severe diaper dermatitis. He was being fed standard infant formula and had watery diarrhea 10–15 times per day. Initial laboratory investigations showed urea 15 mg/dL, creatinine 0.76 mg/dL, AST 36 U/L, ALT 19 U/L, total protein 44 g/L, albumin 28 g/L, sodium 133 mEq/L, chloride 112 mEq/L , potassium 4.2 mEq/L, calcium 8.5 mg/dL, phosphorus 3.5 mg/dL, C-reactive protein 96.3 mg/L, white blood cell count 38,250/µL, hemoglobin 8.6 g/dL, mean corpuscular volume 92.6 fL, and platelet count 386,000/µL. Venous blood gas analysis revealed severe metabolic acidosis (pH 7.14, bicarbonate 10.7 mmol/L, base excess −17.5 mmol/L). Stool microscopy, rotavirus and adenovirus antigen tests, stool cultures and stool reducing substances were negative. Stool electrolyte analysis could not be performed. Fecal elastase level was normal (286 µg/g), and sweat chloride test results were within the normal range. Comprehensive metabolic and immunologic evaluations were unremarkable, including acylcarnitine profile, plasma and urine amino acids, urine organic acids, serum immunoglobulin levels, and lymphocyte subset analysis. CFTR gene analysis was normal. Broad-spectrum antibiotic therapy and intravenous hydration were initiated. Klebsiella pneumoniae was detected in the blood cultures, and the patient was treated for culture-proven sepsis. The elevated inflammatory markers were transient and resolved with supportive treatment, and no evidence of recurrent or persistent systemic inflammatory response was observed during follow-up. As diarrhea did not improve with bowel rest, secretory diarrhea was suspected. Total parenteral nutrition was initiated, and feeding was switched to an amino acid–based formula. Upper gastrointestinal endoscopy and colonoscopy were performed. Endoscopy revealed antral gastritis, bulbitis and duodenitis (Fig. 1), whereas colonoscopy findings were normal. Histopathological examination of gastric biopsies demonstrated Helicobacter pylori–negative chronic gastritis of moderate severity. Duodenal biopsies showed widespread villous flattening with an intraepithelial lymphocyte count of 8 per 100 epithelial cells (Fig. 2). Colonic biopsies revealed normal mucosa. During follow-up, the patient developed recurrent hyponatremia, with sodium levels decreasing to as low as 128 mmol/L, necessitating intravenous correction followed by ongoing oral sodium supplementation, which is still required. Currently, he is fed with an amino acid–based formula, receiving nutrition orally and via intermittent nasogastric tube feeding, and continues oral sodium supplementation. From the age of 4 months onward, the frequency of diarrhea gradually decreased, and bowel movements normalized to 1–2 times per day. However, intermittent abdominal distension and vomiting persisted, necessitating continued partial nasogastric feeding.
At the most recent evaluation at 6 months and 18 days of age, his weight was 6.6 kg (SDS −1.91), length 63 cm (SDS −2.56), and body mass index 16.6 kg/m² (SDS −0.51). Weight-for-length was preserved (SDS −0.33). At 6.5 months of age, he was hospitalized once for bronchiolitis. Whole exome sequencing identified a homozygous frameshift variant in AGR2 (NM_006408.4): c.247_250del (p.His83ValfsTer4). The American College of Medical Genetics and Genomics (ACMG) criteria classifies this variant as likely pathogenic, based on PVS1 and PM2 criteria. Parental testing confirmed heterozygous carrier status for the same variant in both parents, consistent with autosomal recessive inheritance. A heterozygous variant of uncertain significance (VUS) in GUCY2C, which encodes guanylate cyclase C, a receptor involved in intestinal fluid and electrolyte secretion, was also detected in the patient and his asymptomatic father. Although gain-of-function variants in GUCY2C have been associated with autosomal dominant forms of early-onset chronic diarrhea6, its presence in an asymptomatic parent supported its classification as a secondary finding unlikely to explain the clinical phenotype. The clinical features of the patient in comparison with previously reported AGR2-related cases are summarized in Table I. Written informed consent was obtained from the patient’s parents for the publication of this case report.
| GI: gastrointestinal. | |||||||
| Table I. Individual AGR2 variants and associated clinical features reported in the literature and in the present case. | |||||||
| Study | Homozygous AGR2 variant (NM_006408.4) | Age at onset | Gastrointestinal manifestations | Respiratory involvement | Failure to thrive |
Clinical course/outcome (age at last follow-up) |
|
|
Bertoli-Avella et al.5 (9 families, 13 patients) |
Patient 1 | c.211C>A (p.Pro71Thr) exon 4 | 2 wk | None |
Chronic cough, exertional dyspnea, mild bronchiectasis |
Yes, weight below 5th percentile | Childhood |
| Patient 2 | c.211C>A (p.Pro71Thr) exon 4 | 6 mo | None | Chronic cough, mild bronchiectasis | Yes, weight below 5th percentile, height at 10th percentile | Childhood | |
| Patient 3 | c.211C>A (p.Pro71Thr) exon 4 | 1 wk | None | Chronic cough, recurrent wheezing episodes, dyspnea | Yes, weight below 5th percentile | Childhood | |
| Patient 4 | c.349C>T (p.His117Tyr) exon 6 | Birth | Acute gastroenteritis, vomiting, severe gastroesophageal reflux, chronic diarrhea | Chronic cough, pneumonia, hyperactive airway disease | Yes | Childhood | |
| Patient 5 | c.349C>T (p.His117Tyr) exon 6 | 2 d | Chronic diarrhea, episodic vomiting | Mild respiratory tract infections | Yes, weight and height below 5th percentile | Infancy | |
| Patient 6 | c.349C>T (p.His117Tyr) exon 6 | 1 yr | None | Chronic cough, severe pneumonia | Yes | Childhood | |
| Patient 7 | c.330+1G>T, intron 5 | 8 mo | Hepatomegaly | Interstitial lung disease | Yes | Deceased | |
| Patient 8 | c.330+1G>T, intron 5 | 10 d | Choking, vomiting, chronic diarrhea, hepatomegaly | Recurrent wheezing episodes, patch areas of ground glass appearance and scattered consolidations in both lungs | Yes | Childhood | |
| Patient 9 | Large deletion (exon 1-7 chr7:16834456-16918247) | 6 mo | None | Bronchiectasis, chronic cough | Yes | Childhood | |
| Patient 10 | c.349C>T (p.His117Tyr) exon 6 | Birth | Chronic diarrhea (improved after 2 yr), hepatomegaly | Chronic cough, pleural effusion, hilar lymphadenopathy, bronchiectasis | Yes, weight below 3rd percentile | Childhood | |
| Patient 11 | c.349C>T (p.His117Tyr) exon 6 | Birth | Chronic diarrhea | Chronic cough, hilar lymphadenopathy | Yes, weight below 3rd percentile | Early childhood | |
| Patient 12 | c.330+1del, intron 5 | 2 yr | Persistent vomiting, hepatomegaly, persistent cholestasis | Bronchiectasis, chronic cough | Yes | Childhood | |
| Patient 13 | c.428G>A (p.Gly143Glu) exon 7 | 3 d | Chronic diarrhea, abdominal distention with prominent veins | Subsegmental atelectasis | Yes, weight and height below 5th percentile | Early childhood | |
| Al-Shaibi et al.7 | Patient 14 | c.349C>T (p.His117Tyr) | Birth | Chronic diarrhea, upper and lower GI mixed chronic and active cellularity inflammation with clear goblet cell depletion and apoptosis | Bilateral upper chest infiltration with left lower atelectasis | Yes, weight below 3rd percentile | Early childhood |
| Patient 15 | c.349C>T (p.His117Tyr) | Birth | Chronic diarrhea, vomiting, upper and lower GI mixed chronic and active cellularity inflammation with clear goblet cell depletion and apoptosis | Reactive airway disease | Yes | Early childhood | |
| Takada et al.3 | Patient 16 | c.250A>C (p.Ser84Arg) | 4 mo | Gastroesophageal reflux, chronic esophagitis and gastritis, colon biopsy showing crypt distortion, cryptitis, patchy lymphoplasmacytic inflammation, and decreased goblet cells, suggesting ulcerative colitis | Recurrent sinusitis and upper respiratory infections, Bronchiectasis | No | Adolescence |
| Patient 17 | c.250A>C (p.Ser84Arg) | 3 mo | Esophagitis, gastric ulcers and atrophy, and severe pyloric stenosis | Progressive respiratory failure requiring a bi-pulmonary transplantation | Yes, height below 3rd percentile | Adolescence | |
| Present case | Present case | c.247_250del (p.His83ValfsTer4) | 15 d | Chronic diarrhea (improved after 4 mo), vomiting, Upper GI endoscopy showing antral gastritis, bulbitis, and duodenitis, with duodenal biopsies showing villous flattening. | A single bronchiolitis attack | Yes, weight and height below 3rd percentile | Infancy |
Discussion
Congenital diarrheal disorders and enteropathies (CODE) arise from structural and functional defects of absorptive, enteroendocrine, or inflammatory cells within the intestinal epithelium. These defects are determined by mutations in genes expressed throughout the gastrointestinal tract and are most commonly inherited in an autosomal recessive manner. In the first weeks of life, patients affected by CODE typically present with severe diarrhea that can lead to life-threatening dehydration and metabolic acidosis.7 Advances in genomic sequencing have enabled the identification of novel genetic etiologies; however, AGR2-related disease remains extremely rare, with only a limited number of patients reported to date.
Goblet cells express AGR2, a protein disulfide isomerase that is essential for mucus production, which coats the intestinal epithelium and provides protection against infectious and toxic agents.4 The mucus barrier plays a critical role in preventing bacterial invasion and shielding epithelial cells from luminal aggressors, including gastric acid. Mucin 2 (MUC2) is the major gel-forming mucin in the intestine and defective processing or loss of MUC2 leads to impaired mucus barrier integrity and promotes intestinal inflammation.8 While MUC2 is the predominant gel-forming mucin in the intestine, Mucin 5AC (MUC5AC) and Mucin 6 (MUC6) are the principal gel-forming mucins in the stomach, whereas MUC5AC and Mucin 5B (MUC5B) are primarily expressed in the respiratory tract.3,8,9 Recurrent respiratory infections and failure to thrive, with or without diarrhea (RIFTD) results from impaired mucus biosynthesis, and different AGR2 variants affect distinct types of mucus.3,5
Previous studies have highlighted the multisystem nature of AGR2-related disease. Bertoli-Avella et al. reported 13 patients from nine unrelated families with a previously undescribed genetic disorder characterized by recurrent lower respiratory infections, chronic diarrhea, and failure to thrive, a clinical phenotype closely resembling cystic fibrosis. This seminal study provided the first comprehensive evidence linking biallelic AGR2 variants to a cystic fibrosis–like multisystem disorder.5 In another study, Al-Shaibi et al.
investigated siblings with congenital diarrhea who developed severe infantile inflammatory bowel disease due to AGR2 deficiency. Histopathological examination revealed infantile enteropathy, patchy lymphocytic infiltration in the gastric mucosa and extensive intestinal metaplasia characterized by absence of parietal cells and the presence of eosinophilic Paneth-like cells, underscoring the broad gastrointestinal involvement associated with AGR2 dysfunction.8
Our patient exhibited a predominantly gastrointestinal phenotype, characterized by severe congenital secretory diarrhea, failure to thrive, electrolyte imbalance, together with a single episode of bronchiolitis, which cannot be definitively attributed to AGR2-related pulmonary disease but may represent either an early respiratory manifestation or a coincidental common infection of infancy. Longitudinal follow-up and the occurrence of recurrent or persistent respiratory symptoms would be required to establish definite pulmonary involvement in AGR2-related disease in this patient. While gastric biopsies in our case demonstrated Helicobacter pylori–negative chronic gastritis without intestinal metaplasia, duodenal biopsies showed widespread villous flattening, supporting the presence of congenital enteropathy. Although stool electrolyte analysis could not be performed, the persistence of diarrhea despite bowel rest and total parenteral nutrition strongly supported a secretory mechanism in this patient. Notably, diarrhea gradually improved with age, suggesting partial intestinal adaptation, a feature that has been variably reported in previously described patients.5 These findings further support the concept that AGR2-related disease represents a clinical spectrum, in which the predominant organ involvement and disease severity may vary according to the specific AGR2 variant and the affected mucus subtype. The clinical and genetic characteristics of previously reported AGR2-related cases and the present patient are summarized in Table I, highlighting the phenotypic variability of AGR2-related disease.
A notable clinical feature in our patient was recurrent hyponatremia requiring prolonged oral sodium supplementation, despite normal sweat chloride test results and the absence of CFTR mutations. In contrast to cystic fibrosis, in which electrolyte imbalance results from impaired salt reabsorption in sweat glands, we considered that hyponatremia in AGR2-related disease primarily arises from intestinal sodium loss secondary to chronic secretory diarrhea and impaired epithelial barrier function.
The identified homozygous frameshift variant in the AGR2 gene (c.247_250del; p.His83ValfsTer4) is predicted to result in a premature termination codon and subsequent loss of protein function, consistent with the established loss-of-function pathogenic mechanism of biallelic AGR2 variants. Parental carrier testing confirmed autosomal recessive inheritance, further supporting the causal role of this variant in the observed clinical phenotype. The absence of alternative genetic explanations for congenital diarrhea in our patient strengthens the genotype–phenotype correlation.
This case report has several limitations. First, functional studies assessing the direct impact of the identified AGR2 variant on mucus biosynthesis and epithelial barrier function could not be performed. Second, stool electrolyte analysis was not available, which would have provided biochemical confirmation of secretory diarrhea.
In conclusion, this case expands the clinical spectrum of AGR2-related disease by demonstrating a predominantly gastrointestinal presentation characterized by severe congenital secretory diarrhea and electrolyte imbalance requiring prolonged sodium supplementation. Partial clinical improvement over time suggests phenotypic variability and possible intestinal adaptation. Given the rarity of this condition, AGR2 deficiency may be considered in selected infants presenting with severe congenital diarrhea, failure to thrive, electrolyte imbalance, particularly in the setting of consanguinity, even without definite pulmonary involvement.
Ethical approval
Written informed consent was obtained from the patient’s parents for the publication of this case report.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Younis M, Rastogi R, Chugh A, Rastogi S, Aly H. Congenital diarrheal diseases. Clin Perinatol 2020; 47: 301-321. https://doi.org/10.1016/j.clp.2020.02.007
- Gaibee Z, Warner N, Bugda Gwilt K, et al. The genetic architecture of congenital diarrhea and enteropathy. N Engl J Med 2025; 392: 1297-1309. https://doi.org/10.1056/NEJMoa2405333
- Takada S, Gallo S, Silva S, et al. A novel AGR2 variant causing aberrant monomer-dimer equilibrium leading to severe respiratory and digestive symptoms. J Clin Immunol 2024; 45: 55. https://doi.org/10.1007/s10875-024-01847-x
- Park SW, Zhen G, Verhaeghe C, et al. The protein disulfide isomerase AGR2 is essential for production of intestinal mucus. Proc Natl Acad Sci U S A 2009; 106: 6950-6955. https://doi.org/10.1073/pnas.0808722106
- Bertoli-Avella A, Hotakainen R, Al Shehhi M, et al. A disorder clinically resembling cystic fibrosis caused by biallelic variants in the AGR2 gene. J Med Genet 2022; 59: 993-1001. https://doi.org/10.1136/jmedgenet-2021-108150
- Wolfe RM, Mohsen AW, Walsh Vockley C, et al. Novel GUCY2C variant causing familial diarrhea in a Mennonite kindred and a potential therapeutic approach. Am J Med Genet A 2021; 185: 2046-2055. https://doi.org/10.1002/ajmg.a.62207
- Berni Canani R, Terrin G, Cardillo G, Tomaiuolo R, Castaldo G. Congenital diarrheal disorders: improved understanding of gene defects is leading to advances in intestinal physiology and clinical management. J Pediatr Gastroenterol Nutr 2010; 50: 360-366. https://doi.org/10.1097/MPG.0b013e3181d135ef
- Al-Shaibi AA, Abdel-Motal UM, Hubrack SZ, et al. Human AGR2 deficiency causes mucus barrier dysfunction and infantile inflammatory bowel disease. Cell Mol Gastroenterol Hepatol 2021; 12: 1809-1830. https://doi.org/10.1016/j.jcmgh.2021.07.001
- Schroeder BW, Verhaeghe C, Park SW, et al. AGR2 is induced in asthma and promotes allergen-induced mucin overproduction. Am J Respir Cell Mol Biol 2012; 47: 178-185. https://doi.org/10.1165/rcmb.2011-0421OC
Copyright and license
Copyright © 2026 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









