
Graphical Abstract
Abstract
Background. The α-actinin-4 (ACTN4) gene encodes an actin-binding protein, which plays a crucial role in maintaining the structure and function of podocytes. Previous studies have confirmed that ACTN4 mutations can lead to focal segmental glomerulosclerosis-1 (FSGS1), a rare disease primarily manifesting in adolescence or adulthood, characterized by mild to moderate proteinuria, with some cases progressing slowly to end-stage renal disease.
Case Presentation. We report a 12.5-year-old boy who presented with non-nephrotic range proteinuria, hyperuricemia, markedly reduced bilateral kidney volume, and stage 3 chronic kidney disease (CKD). An ophthalmic examination revealed optic disc dysplasia in the right eye. The results of whole-exome sequencing revealed a de novo variant in the ACTN4, a previously unreported variant.
Conclusions. We reported a novel sporadic ACTN4 variant and reviewed previously reported cases. Through analysis of the genotypes and clinical phenotypes of reported cases, we found that ACTN4 variants may not always present as FSGS1, and there was significant phenotypic heterogeneity among individuals. Notably, mutations affecting residues 260-265 are associated with collapsing glomerulopathy and rapid progression to end-stage kidney disease in prior studies, whereas the p.Ala278del variant in our case, located outside this region, exhibited stable CKD3. This suggests domain-specific genotype-phenotype correlations. However, this association requires further validation through additional cases and experiments. Our findings may have significant implications for clinical diagnosis, prognosis assessment, and scientific research on kidney diseases related to ACTN4 variants.
Keywords: ACTN4, variant, focal segmental glomerulosclerosis, proteinuria, eye
Introduction
With the continuous development of genomics research, a deeper understanding has been gained of renal diseases caused by monogenic inheritance. Research has found that up to 30% of patients with steroid-resistant nephrotic syndrome are caused by monogenic diseases.1 The α-actinin-4 (ACTN4) gene is located on human chromosome 19 and is involved in encoding the cytoskeleton and actin-binding protein ACTN4. It is crucial for maintaining podocytes’ normal structure and function. Additionally, variants in ACTN4 have been confirmed as a monogenic cause of steroid-resistant nephrotic syndrome.2,3
It has been confirmed that mutations in ACTN4 can lead to autosomal dominant focal segmental glomerulosclerosis-1 (FSGS1), a rare disease with a relatively high probability of familial occurrence. Onset usually occurs during adolescence or later and is characterized by mild to moderate proteinuria accompanied by renal dysfunction, with some cases progressing slowly to end-stage kidney disease (ESKD).2 Currently, there are approximately 107 reported cases of renal disease caused by ACTN4 mutations, including 28 mutation types.1-16 Since Kaplan et al. first reported in 2000, it has been generally considered that ACTN4 mutations are associated with FSGS1. However, in recent years, there have been reports of sporadic ACTN4 variants.4-6,9 Here, we report a case of sporadic ACTN4 variant causing chronic kidney disease (CKD) in a child. The variant in this case has not been reported previously, and this patient is currently the only reported case with concurrent ocular lesions. Our case report and literature review may have expanded the phenotypic and genotypic spectrum associated with ACTN4 variants. We also identified a potential correlation between genotype and phenotype. Moreover, some of our findings require further validation through additional cases and functional studies, thereby providing new directions for future research on ACTN4-related disorders.
Case Presentation
A 12.5-year-old boy presented to our hospital due to persistently elevated serum creatinine and uric acid levels over one year, during which no specific treatment was administered. He did not present any manifestations such as hematuria, foamy urine, enuresis, oliguria, urinary frequency, urgency, edema, rash, or pallor. The patient had no significant past medical history. His father had hyperuricemia managed with febuxostat, though specific pre-treatment uric acid levels were unavailable due to incomplete medical records. No abnormalities were found in the father’s urine routine examination, and his estimated glomerular filtration rate (eGFR), calculated using the CKD-EPI equation, was within the normal range (90-120 mL/min/1.73 m²). Moreover, there is no history of consanguineous marriage or other family history. There were no apparent abnormalities in the boy’s physical examination. A 24-hour proteinuria of 9.5 mg/m²/hour was detected, consistent with non-nephrotic range proteinuria. Renal function tests showed elevated serum creatinine (maximum 1.5mg/dL) and uric acid (maximum 11.6 mg/dL) levels. The patient’s eGFR, calculated using the Schwartz formula, was 59 mL/min/1.73 m², consistent with CKD stage 3a according to the KDIGO 2024 Clinical Practice Guideline.17 Ultrasonography of the urinary system revealed significant bilateral renal volume reduction (right kidney 7.7 × 3.5 × 3.1 cm, left kidney 8.3 × 3.8 × 3.6 cm) with enhanced echogenicity of the renal parenchyma. Ophthalmic examination revealed refractive errors and right optic disc dysplasia. The patient exhibited no clinical features suggestive of systemic lupus erythematosus (SLE). Serological testing showed no evidence of SLE-associated autoantibodies or hypocomplementemia. Screening tests for blood lipids, blood glucose, serum albumin, coagulation function, immunological disorders, and infectious diseases (such as hepatitis B, tuberculosis, and human immunodeficiency virus) showed no abnormalities. Malignancy was excluded through tumor markers and chest-abdomen-pelvis computed tomography scans.
Considering the insidious onset of the patient’s condition and the absence of apparent triggers leading to CKD3, along with his father’s hyperuricemia, further genetic examination was conducted. Genomic DNA was extracted from the proband and parents using the Qiagen Blood DNA Mini Kit (Qiagen, Germany). Whole-exome sequencing (WES) was performed by MyGenostics (Beijing, China) using the GenCap® Exome Enrichment V6.0 probe (51 Mb target region, covering ~23,000 genes). Sequencing was conducted on an Illumina NovaSeq 6000 platform with 200× mean coverage depth, ensuring >99% of target regions achieved ≥20× coverage. Raw reads were filtered using CutAdapter to remove low-quality sequences. Clean reads were aligned to the GRCh37/hg19 reference genome using BWA. GATK was used for single nucleotide variation and inserts and deletions. Variants were annotated using ANNOVAR and filtered against population databases (1000 Genomes, Exome Variant Server, ExAC) with a minor allele frequency <0.1%. SIFT, PolyPhen2, MutationTaster, and GERP were used for in silico analysis.
WES revealed a novel heterozygous variant in ACTN4 (NM_004924.6:c.832_834del, p.Ala278del). In silico tools (SIFT, PolyPhen2, MutationTaster, and GERP) predicted uncertain significance for the p.Ala278del variant. The variant was highly associated with the patient’s clinical phenotype, and no other variants related to the patient’s phenotype were detected. His parents do not have variants at this locus or any other variants. The variant is considered to be a de novo variant in the patient (Fig. 1). According to the standards of the American College of Medical Genetics and Genomic, this variant is classified as likely pathogenic (PS2+PM2_Supporting+PM4).18
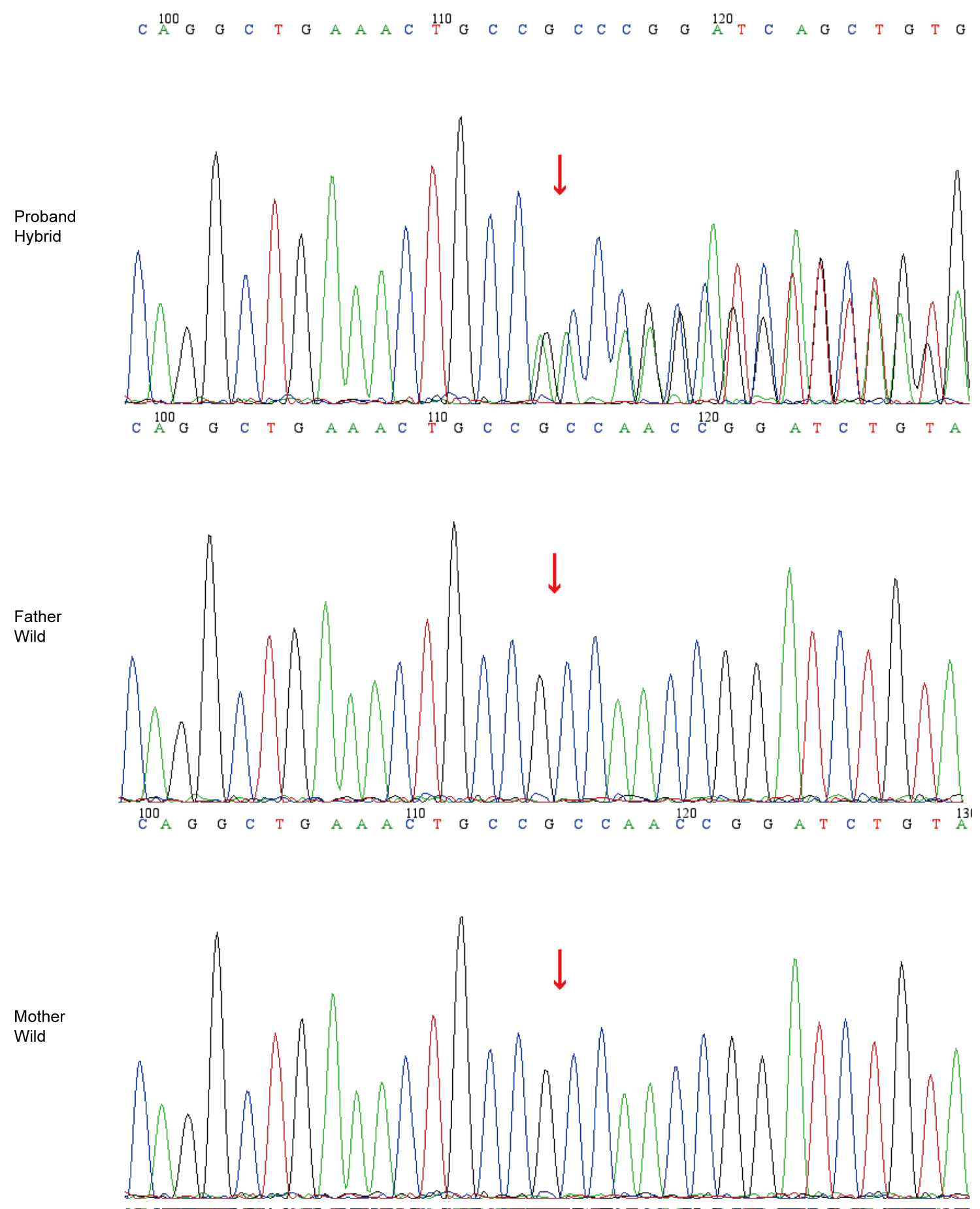
Over 18 months of follow-up, the patient’s renal function remained stable with persistent non-nephrotic proteinuria. No corticosteroids or immunosuppressive agents were administered, as clinical progression was indolent and genetic testing indicated an ACTN4 variant associated with steroid resistance. Informed consent for publication was obtained from the patient’s legal guardian.
Discussion
Podocytes serve as critical structures maintaining the renal filtration barrier. Mutations in ACTN4 can cause podocyte dysfunction, thereby initiating the development of CKD. Kos et al. found that knockout mice lacking the ACTN4 showed disappearance of foot processes and reduced expression of ACTN4 in podocytes. Most knockout mice died around the perinatal period while surviving mice exhibited proteinuria and FSGS.19 This indicates the crucial role of ACTN4 in renal function. Actin in podocytes is crucial for the construction and maintenance of the cytoskeleton. ACTN4 encoded by the ACTN4 is an actin-binding protein primarily localized to the foot processes, playing a significant role in regulating the actin cytoskeleton of podocytes. Studies have indicated that mutations in ACTN4 lead to the mislocalization of the encoded protein and the formation of intracellular aggregates. This results in instability of the formed ACTN4, increased affinity with actin, and reduced dissociation and degradation rates, ultimately disrupting the actin cytoskeleton in podocytes.2,3,6,11,20 Consequently, mutations in ACTN4 disrupt podocytes’ normal structure and function, causing renal disease.
We reviewed previously reported and clinically significant cases with ACTN4 variants (Supplementary Table I). A systematic search of PubMed and Google Scholar was performed using the terms “ACTN4 AND mutation” and “ACTN4 AND variant”. Articles were restricted to those published in the English language. Studies were included if they (1) confirmed ACTN4 variants via genetic testing, (2) provided renal manifestations, and (3) excluded non-human or phenotype-unclear cases. A total of 107 cases (including ours) were eligible for analysis. Previously, our understanding of renal disease caused by ACTN4 variants mainly focused on the development of FSGS1, characterized by familial aggregation, and later onset (mainly in adolescents or adults), with some cases progressing slowly to ESKD.2,3 However, through a comprehensive review of the clinical characteristics of previously reported cases, particularly considering some newer reports in recent years involving phenotypes different from those previously described, we found that ACTN4 variants may not solely manifest as FSGS1, and there is significant heterogeneity in individual clinical phenotypes. It mainly includes the following aspects: 1. Variants may not always be inherited within families. Among the 107 reported cases: 18 were sporadic; 3 had unknown family history; and the remaining 86 were familial.1,4-6,9-13 The 18 sporadic cases had no family history of renal disease, with onset age ranging from 3.7 to 17 years old, mainly presenting with proteinuria or nephrotic syndrome, almost all progressing to ESKD. 2. Some patients developed the disease before adolescence (10 cases occurred before adolescence, with the youngest onset age being three years old).3,5-7,9,11 3. There were patients with rapid progression (nine cases progressed to ESKD within three years of onset, with one case progressing to ESKD within six months). 4. Patients with childhood-onset disease progressed to ESKD more rapidly than those with adult-onset disease.1,3,4,6,8-10,12 However, the mechanisms underlying the heterogeneity of clinical phenotypes remain unclear and warrant further investigation.
ACTN4 comprises three main structural domains: 1. The N-terminal actin-binding domain (ABD), which comprises two calponin-homology (CH) domains; 2. The spectrin domain (composed of four repeat sequences); 3. A pair of C-terminal EF-hands.21 We have listed the amino acid sequence positions on the ACTN4 protein corresponding to the reported variant sites (Fig. 2). It can be observed that variants reported so far are more common in the ABD region. At the same time, there are fewer reports of mutations in the spectrin domain. To date, six cases of mutations in the spectrin domain have been reported, all in Asian populations: five Chinese (including the present case) and one Japanese.5,15 It suggests that the spectrin domain may be a hotspot variant region in Asian populations. While this clustering could suggest ethnic-specific factors, current evidence is insufficient to confirm a biological basis. Larger multi-ethnic cohorts and functional studies are needed to distinguish technical biases (e.g., regional diagnostic practices) from true genetic predisposition.
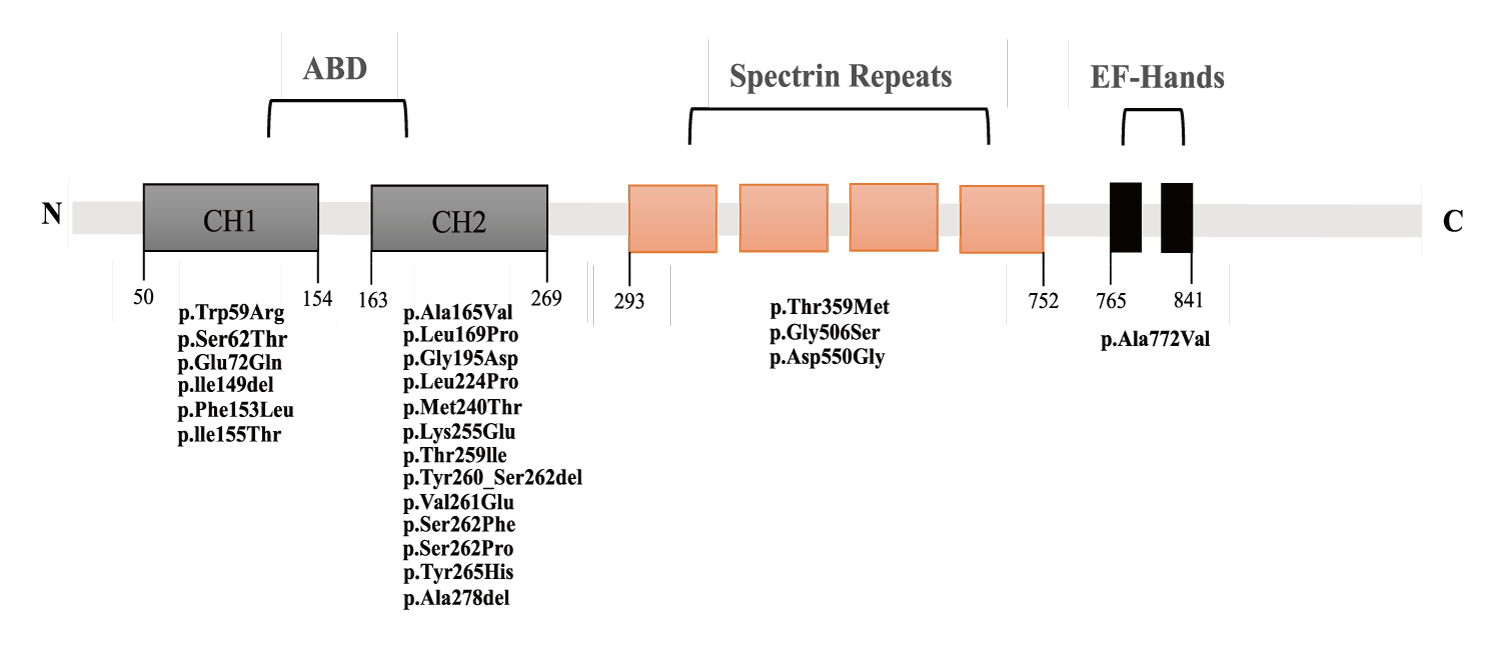
Review of reported ACTN4 mutations (Supplementary Table I) reveals a potential genotype-phenotype correlation: Pathogenic mutations within residues 260-265 (e.g., p.Ser262Phe, p.Val261Glu, p.Tyr265His, p.Tyr260_Ser262del [denoting deletion of residues Tyr260 through Ser262]; cases 9, 11, 13, 14, 21, 25, 30 in Supplementary Table I) are strongly associated with collapsing glomerulopathy and rapid progression to ESKD (0.5-3 years), as documented in prior studies.1,6-9,12 For example, Kakajiwala et al. described a child with p.Ser262Phe who progressed to ESKD within six months6, while Feng et al. reported a p.Tyr265His variant linked to collapsing lesions and ESKD in six months.12 In contrast, our patient’s novel p.Ala278del variant, located outside this region, presented with stable CKD3 over 18 months, suggesting that mutation position may modulate phenotypic severity. We hypothesize that the 260-265 region, which overlaps with the actin-binding domain, may be critical for cytoskeletal stability, and variants here could disrupt actin dynamics more severely than those in other domains. This effect likely arises because slower degradation of ACTN4-actin complexes in these variants leads to abnormal protein clumps inside podocytes. These aggregates destabilize the cytoskeleton, accelerating disease progression.11 However, confirming such an association between genotype and phenotype necessitates further investigation with additional cases and experimental validation in the future. Moreover, the limited clinical data on mutations in other domains (e.g., spectrin domain) precludes a systematic comparison of domain-specific phenotypic differences. Future studies with larger cohorts and functional validation are needed to confirm whether mutation location directly dictates clinical severity.
Due to the similarity in development, structure, and physiology between the kidneys and eyes, ocular involvement is relatively common in hereditary renal diseases.22,23 To our knowledge, this is the first reported case associating an ACTN4 variant with ocular manifestations. Retina, lens, and ocular muscle abnormalities have been observed in mouse models with ACTN4 variants.24 Human Protein Atlas showing low but detectable ACTN4 expression in human retina. While ACTN4 is primarily characterized in podocytes, its potential role in ocular tissues remains speculative. This case highlights the phenotypic expansion of ACTN4-related disorders, though the causal link between the variant and ocular manifestations has yet to be established. Future studies could validate the ocular localization of ACTN4 and explore whether this variant exerts pleiotropic effects beyond the kidney.
Previously reported cases of ACTN4 variants with clinical treatment outcomes, except for two cases with variants occurring at splice sites5, all were resistant to steroids. In the patient Odenthal et al. reported, proteinuria was significantly alleviated upon initial use of cyclosporine. It may be attributed to the impact of such immunosuppressive agents on the cytoskeleton and attenuation of immune response. The study suggests that cyclosporine may help delay renal replacement therapy or renal transplantation.11 While this suggests a potential role for cytoskeletal modulation, its applicability to ACTN4-related nephropathy remains unproven. Importantly, cyclosporine’s nephrotoxicity risk necessitates cautious patient selection. Further studies are needed to define its utility in ACTN4-related disorders.
As the significantly reduced bilateral kidney volume observed in the patient upon presentation and the heightened risk of renal biopsy-related bleeding, coupled with the manifestation of sclerosing nephropathy in most cases, renal biopsy was not performed in the case we reported. The patient presented with persistent proteinuria accompanied by renal insufficiency without a significant family history. However, genetic testing revealed a novel variant in the ACTN4. It suggests that patients with unexplained CKD may harbor genetic abnormalities despite lacking a clear family history of renal disease. Therefore, physicians should not refrain from genetic testing simply because patients lack a family history of renal disease. For patients who cannot undergo renal biopsy, genetic testing not only aids in making a definitive diagnosis but also guides treatment and facilitates planning for future renal transplantation. Our summary of previous cases shows that the majority of patients were steroid-resistant. Additionally, the probability of monogenic diseases in steroid-resistant nephrotic syndrome patients is not low. Early genetic testing for such patients can help avoid excessive use of corticosteroids. Hence, genetic testing methods are necessary to assist clinicians in disease management.
This study has limitations, as functional validation of the p.Ala278del variant was not conducted. Moreover, the variant’s uncertain prediction by in silico tools likely stems from their limited training data for in-frame deletions. However, the following aspects might support its pathogenicity: this variant is de novo and has not been detected in the parents or population databases; the patient’s clinical presentation is consistent with ACTN4-related nephropathies; and this variant meets the criteria for ‘likely pathogenic’ (PS2+PM2_Supporting+PM4). Future studies could prioritize in vitro assays and structural modeling to confirm its pathogenic mechanism.
To the best of our knowledge, this is the first clinical report of a pediatric patient harboring a novel sporadic ACTN4 variant presenting with CKD3 and concurrent ocular involvement. While murine models and low-level retinal ACTN4 expression suggest plausible biological links between ACTN4 dysfunction and ocular phenotypes, the causal relationship in humans remains speculative. This case highlights the potential phenotypic expansion of ACTN4-related disorders and underscores the need for functional studies to validate ocular pathogenicity mechanisms. Moreover, our literature review reveals phenotypic heterogeneity in ACTN4-related nephropathies and potential genotype-phenotype correlation. These findings are essential for clinical diagnosis, treatment, and prognosis assessment and provide direction for future research on this disease.
Ethical approval
Informed consent was obtained from the patient’s legal guardian for publication.
Source of funding
The authors declare that the study is funded by: 1. Chen Xiaoping Science and Technology Development Foundation’s 2021 Immunological Disease Research, grant number: CXPJJH121002-202107; 2. Recent Efficacy and Safety Study of Immunoadsorption Therapy for Severe Active Systemic Lupus Erythematosus in Children, grant number: 20H0094DNA.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Bierzynska A, McCarthy HJ, Soderquest K, et al. Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int 2017; 91: 937-947. https://doi.org/10.1016/j.kint.2016.10.013
- Kaplan JM, Kim SH, North KN, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet 2000; 24: 251-256. https://doi.org/10.1038/73456
- Weins A, Kenlan P, Herbert S, et al. Mutational and biological analysis of alpha-actinin-4 in focal segmental glomerulosclerosis. J Am Soc Nephrol 2005; 16: 3694-3701. https://doi.org/10.1681/ASN.2005070706
- He Z, Wu K, Xie W, Chen J. Case report and literature review: a de novo pathogenic missense variant in ACTN4 gene caused rapid progression to end-stage renal disease. Front Pediatr 2022; 10: 930258. https://doi.org/10.3389/fped.2022.930258
- Meng L, Cao S, Lin N, et al. Identification of a novel ACTN4 gene mutation which is resistant to primary nephrotic syndrome therapy. Biomed Res Int 2019; 2019: 5949485. https://doi.org/10.1155/2019/5949485
- Kakajiwala AK, Meyers KE, Bhatti T, Kaplan BS. Rapid progression to end-stage renal disease in a child with a sporadic ACTN4 mutation. Clin Nephrol Case Stud 2015; 3: 14-18. https://doi.org/10.5414/CNCS108616
- Choi HJ, Lee BH, Cho HY, et al. Familial focal segmental glomerulosclerosis associated with an ACTN4 mutation and paternal germline mosaicism. Am J Kidney Dis 2008; 51: 834-838. https://doi.org/10.1053/j.ajkd.2008.01.018
- Pollak MR, Alexander MP, Henderson JM. A case of familial kidney disease. Clin J Am Soc Nephrol 2007; 2: 1367-1374. https://doi.org/10.2215/CJN.02040507
- Giglio S, Provenzano A, Mazzinghi B, et al. Heterogeneous genetic alterations in sporadic nephrotic syndrome associate with resistance to immunosuppression. J Am Soc Nephrol 2015; 26: 230-236. https://doi.org/10.1681/ASN.2013111155
- Bartram MP, Habbig S, Pahmeyer C, et al. Three-layered proteomic characterization of a novel ACTN4 mutation unravels its pathogenic potential in FSGS. Hum Mol Genet 2016; 25: 1152-1164. https://doi.org/10.1093/hmg/ddv638
- Odenthal J, Dittrich S, Ludwig V, et al. Modeling of ACTN4-based podocytopathy using Drosophila nephrocytes. Kidney Int Rep 2022; 8: 317-329. https://doi.org/10.1016/j.ekir.2022.10.024
- Feng D, Steinke JM, Krishnan R, Birrane G, Pollak MR. Functional validation of an Alpha-Actinin-4 mutation as a potential cause of an aggressive presentation of adolescent focal segmental glomerulosclerosis: implications for genetic testing. PLoS One 2016; 11: e0167467. https://doi.org/10.1371/journal.pone.0167467
- Dai S, Wang Z, Pan X, et al. ACTN4 gene mutations and single nucleotide polymorphisms in idiopathic focal segmental glomerulosclerosis. Nephron Clin Pract 2009; 111: c87-c94. https://doi.org/10.1159/000191198
- Barua M, Brown EJ, Charoonratana VT, Genovese G, Sun H, Pollak MR. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int 2013; 83: 316-322. https://doi.org/10.1038/ki.2012.349
- Nagano C, Hara S, Yoshikawa N, et al. Clinical, pathological, and genetic characteristics in patients with focal segmental glomerulosclerosis. Kidney360 2022; 3: 1384-1393. https://doi.org/10.34067/KID.0000812022
- Kalmár T, Turkevi-Nagy S, Bitó L, et al. Phenotype-genotype correlations in three different cases of adult-onset genetic focal segmental glomerulosclerosis. Int J Mol Sci 2023; 24: 17489. https://doi.org/10.3390/ijms242417489
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2024 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int 2024; 105(4 Supplement): S117-S314. https://doi.org/10.1016/j.kint.2023.10.018
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405-424. https://doi.org/10.1038/gim.2015.30
- Kos CH, Le TC, Sinha S, et al. Mice deficient in alpha-actinin-4 have severe glomerular disease. J Clin Invest 2003; 111: 1683-1690. https://doi.org/10.1172/JCI17988
- Yao J, Le TC, Kos CH, et al. Alpha-actinin-4-mediated FSGS: an inherited kidney disease caused by an aggregated and rapidly degraded cytoskeletal protein. PLoS Biol 2004; 2: e167. https://doi.org/10.1371/journal.pbio.0020167
- Lee SH, Weins A, Hayes DB, Pollak MR, Dominguez R. Crystal structure of the actin-binding domain of alpha-actinin-4 Lys255Glu mutant implicated in focal segmental glomerulosclerosis. J Mol Biol 2008; 376: 317-324. https://doi.org/10.1016/j.jmb.2007.11.084
- Farrah TE, Dhillon B, Keane PA, Webb DJ, Dhaun N. The eye, the kidney, and cardiovascular disease: old concepts, better tools, and new horizons. Kidney Int 2020; 98: 323-342. https://doi.org/10.1016/j.kint.2020.01.039
- Booij JC, Baas DC, Beisekeeva J, Gorgels TG, Bergen AA. The dynamic nature of Bruch’s membrane. Prog Retin Eye Res 2010; 29: 1-18. https://doi.org/10.1016/j.preteyeres.2009.08.003
- Zhu V, Huang T, Wang D, Colville D, Mack H, Savige J. Ocular manifestations of the genetic causes of focal and segmental glomerulosclerosis. Pediatr Nephrol 2024; 39: 655-679. https://doi.org/10.1007/s00467-023-06073-y
Copyright and license
Copyright © 2025 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









