
Abstract
Background. Diffuse leptomeningeal glioneuronal tumors (DLGNTs) are rare pediatric central nervous system neoplasms with poorly defined treatment strategies and prognostic factors. Although chemotherapy (CHT) is frequently used, the role of radiotherapy (RT), particularly craniospinal irradiation (CSI), remains unclear.
Case Presentations. We present a case series of three pediatric patients diagnosed with DLGNTs and treated with CSI at an initial dose of 36 Gy, with an additional boost to 54 Gy. Patients were evaluated for early radiological response post-CSI and its potential prognostic implications, alongside their clinical and histological features. Two patients demonstrated significant radiological regression after 36 Gy of CSI, with continued improvement 1.5 months post-treatment. These patients remained stable for 88 and 27 months, respectively, without further disease progression. The third patient exhibited disease progression despite CSI and concurrent temozolomide, ultimately succumbing to the disease within 10 months. Notably, this patient had a Ki-67/MIB-1 index of 70%, while surviving patients had lower proliferation indices.
Conclusions. Our findings suggest that an early favorable response to 36 Gy of CSI may serve as a prognostic indicator in DLGNTs. This study highlights the potential value of CSI in managing these tumors and underscores the need for further research to establish standardized treatment approaches.
Keywords: craniospinal irradiation, diffuse leptomeningeal glioneuronal tumor, pediatrics, central nervous system neoplasms, prognosis
Introduction
Diffuse leptomeningeal glioneuronal tumors (DLGNTs), previously known as primary diffuse leptomeningeal oligodendroglioma or disseminated oligodendroglial-like leptomeningeal tumors of childhood, have gained recognition over the past decade.1-3 First introduced in the 2016 World Health Organization (WHO) classification, DLGNTs are now included in the 2021 WHO classification under glioneuronal and neuronal tumors.4 Although some molecular alterations associated with the disease such as KIAA1549-BRAF fusion and 1p deletion have been identified, a formal grading system for these tumors has yet to be established2,3, and additional molecular changes are still under investigation.5
DLGNTs are more commonly observed in the pediatric population.4 Patients often present with hydrocephalus and may require ventriculoperitoneal (VP) shunt surgery.6 Other symptoms include headache, confusion, ataxia, focal neurological deficits, and symptoms related to spinal cord compression.7 Typical magnetic resonance imaging (MRI) findings in DLGNT include diffuse leptomeningeal enhancement along the surfaces of the brain and spinal cord, often described as a “sugar-coating” or “cobweb-like” pattern.5 T2-hyperintense cystic or nodular deposits are frequently found along the parenchymal surface, particularly in areas such as the cerebellum, brainstem, temporal lobes near the Sylvian fissures, hippocampi, and medial occipital lobes.3,5
DLGNT lacks established treatment guidelines, with approaches varying across cases.8-10 Surgical resection is mainly for biopsy or symptom relief, not cure, due to the disease’s disseminated nature.8,10 Chemotherapy (CHT) is first-line, but responses are often partial or stable.8 Radiotherapy (RT) has unclear efficacy, with mixed outcomes reported9,11-14, and is typically reserved for progressive disease, especially in pediatric patients, due to potential side effects.1,3,6,8,15 Given DLGNT’s slow progression, RT is rarely used initially.
Here, we aim to present three cases of DLGNT treated with craniospinal irradiation (CSI) as the primary approach, with an additional tumor bed boost. This is anticipated to be the first publication of its kind in literature.
Case Presentations
The present study was approved by the Ege University Medical Research Ethics Committee (approval number: 24-8T/52). All participants provided informed consent. The three cases of DLGNT presented here were managed at the Ege University Faculty of Medicine, Department of Radiation Oncology between 2017 and 2023. They were treated with craniospinal irradiation (CSI) as the primary approach, with an additional tumor bed boost, reaching a cumulative dose of 54 Gy. Radiological response to CSI was assessed based on the Response Assessment in Neuro-Oncology (RANO) criteria, which evaluates changes in contrast enhancement, non-enhancing lesions, and clinical status.16 CHT was specifically selected for the patient based on the tumor board’s decision. We evaluated the patients’ responses to treatment and believe that the early favorable response to 36 Gy of CSI carries a potential prognostic significance.
The overview of clinical presentations and interventions for 3 cases of DLGNTs is summarized in Supplementary Table S1.
Case 1
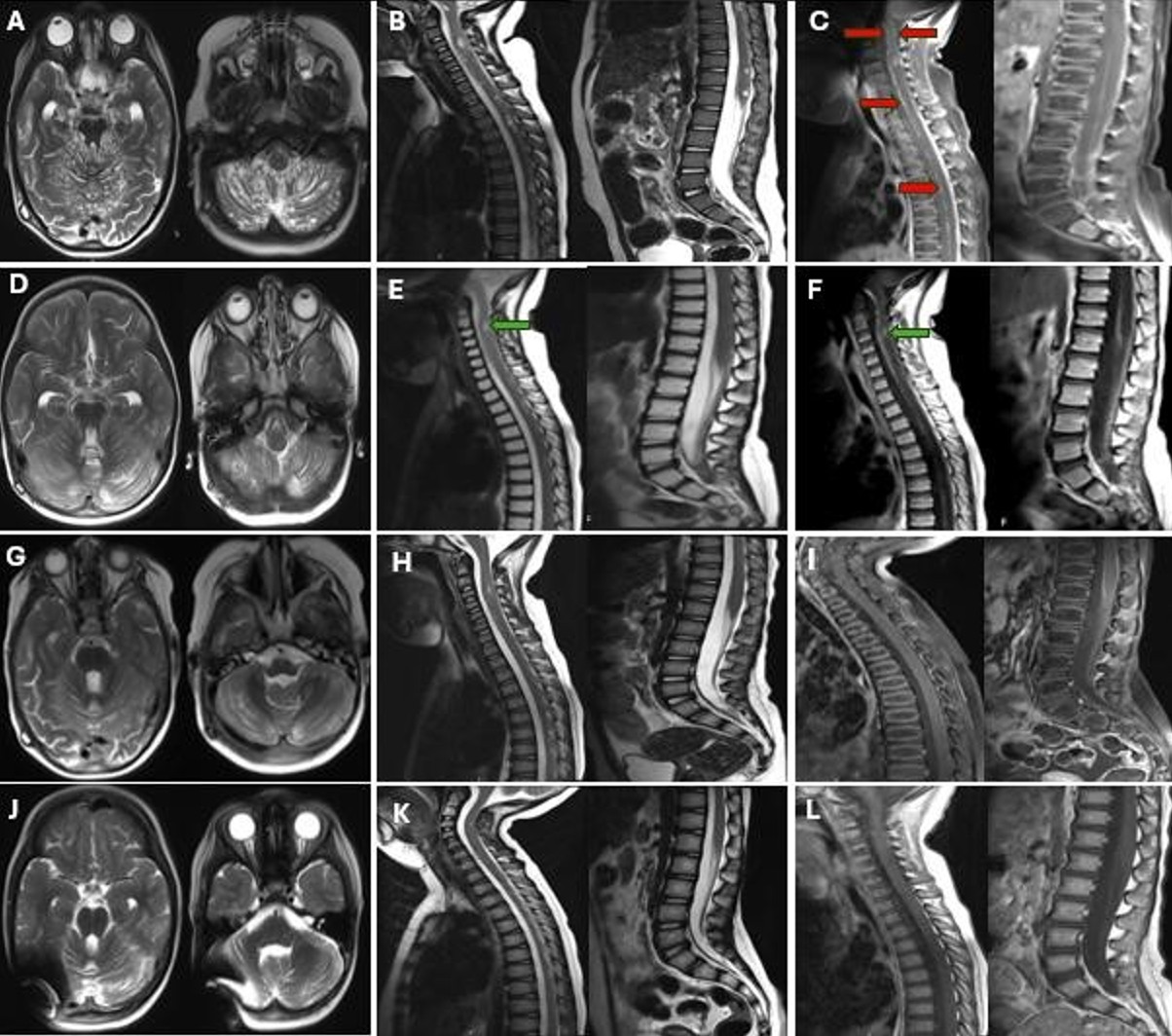
A 3-year-old girl was referred to our hospital with a 5-month history of imbalance and gait disturbances. During the neurological examination, she presented with lethargy, bilateral nystagmus, ataxia, and exaggerated deep tendon reflexes. A computed tomography (CT) scan revealed hydrocephalus, cisternal dilatation, and hypodense nodular lesions in the posterior fossa. VP shunt was performed, and a cerebrospinal fluid (CSF) sample was collected. - CSF analysis was unremarkable. Subsequent cranial MRI revealed extensive T2-hyperintense cystic lesions along the subarachnoid spaces in the posterior fossa, as well as in the inferior frontal and temporal lobes (Fig. 1A). Spinal MRI exhibited similar T2-hyperintense and T1-hypointense cystic lesions, accompanied by leptomeningeal thickening and enhancement (Fig. 1B, Fig. 1C).
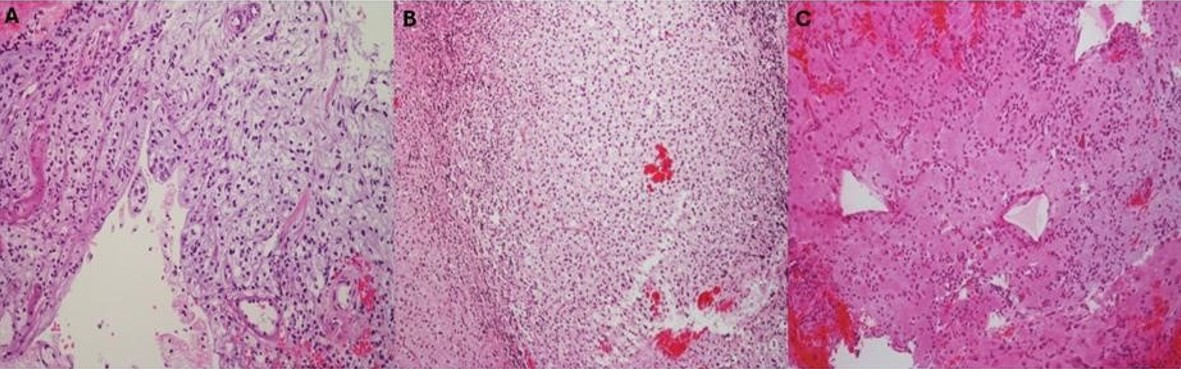
A cerebellar biopsy was performed. Histopathological examination revealed low to moderate cellularity, consisting of uniform tumor cells (Fig. 2A). Immunohistochemistry was consistent with DLGNT, with a Ki-67/MIB-1 index of 10% (Supplementary Table S2).
The patient received RT (36 Gy CSI and 54 Gy to the tumor bed) without concurrent CHT, showing significant cranial and spinal lesion regression on MRI (Fig. 1D-I). Post-RT, she underwent carboplatin and vincristine chemotherapy for 6 years and 3 months, which was stopped due to stable disease. She has remained off treatment and stable for the past 8 months, with a total survival of 88 months (Fig. 1J-L). Follow-up revealed short stature and mild periventricular leukoencephalopathy, but no other clinical or neurological issues.
Case 2
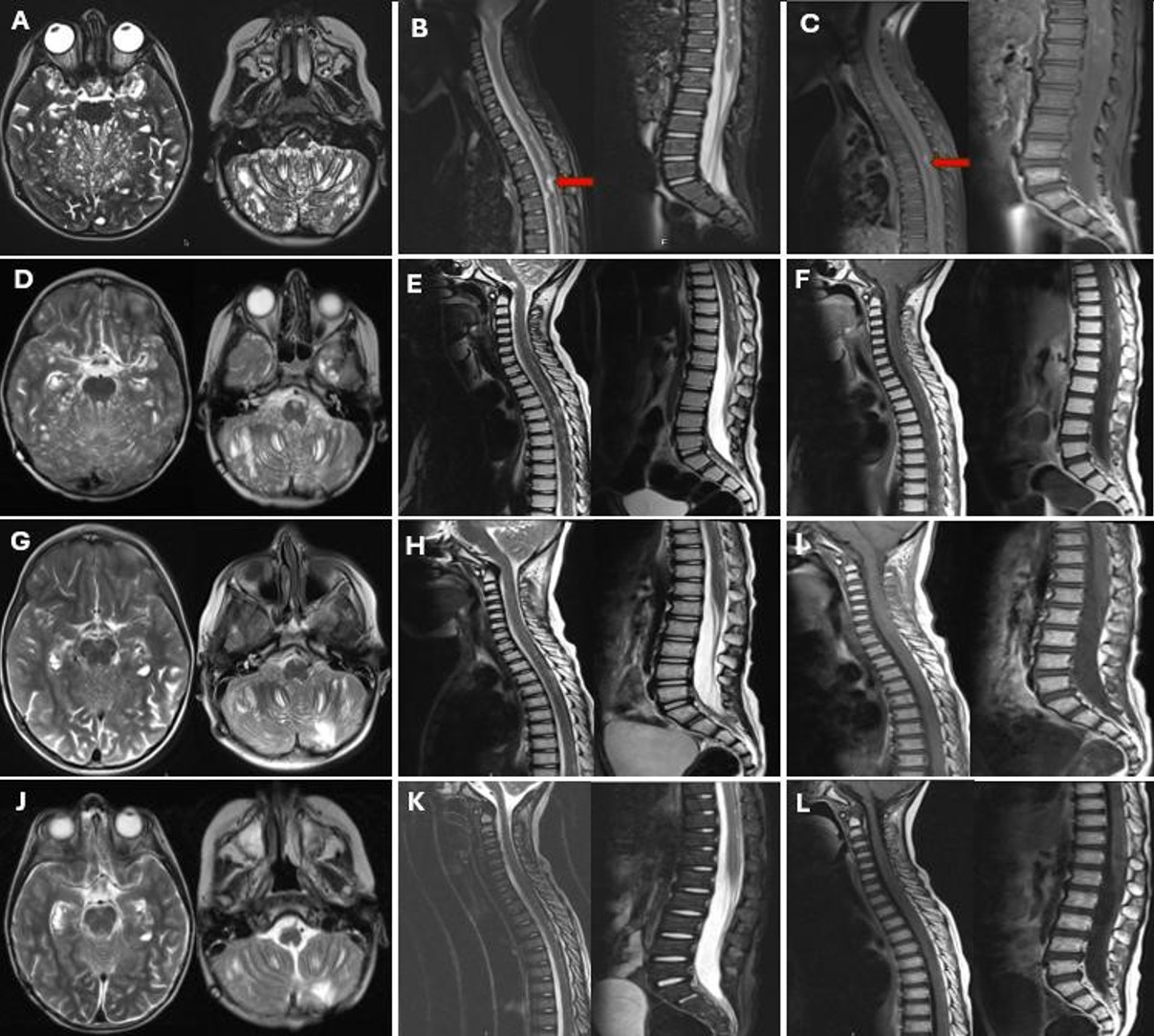
A 4-year-old boy presented to our hospital with symptoms of seizure, lethargy, nausea, and vomiting, which had started 1.5 months prior to admission. Neurological examination revealed inward deviation of the right eye and bilateral lower extremity weakness. It was learned that he had been diagnosed with hydrocephalus and underwent VP shunt placement at the age of 3, approximately one year ago. The cranial MR images from one year ago showed no additional findings aside from hydrocephalus. Current imaging shows no signs of VP shunt dysfunction. Current MRI demonstrated multiple T1 hypointense/T2 hyperintense cystic lesions in the posterior fossa, frontal, and temporal lobes, with leptomeningeal thickening and enhancement in the brainstem, cerebellum, and hemispheres (Fig. 3A). Spinal MRI revealed diffuse leptomeningeal enhancement and multicystic lesions, the largest 1 cm at T5 (Fig. 3B, Fig. 3C).
Cerebellar biopsy showed oligodendrocyte-like cells with perinuclear halos with low to moderate cellularity (Fig. 2B). Immunohistochemistry was consistent with DLGNT (Supplementary Table S2), with a Ki-67/MIB-1 proliferation index of 1%, and benign CSF cytology.
Radiotherapy (36 Gy craniospinal irradiation and 54 Gy to the tumor bed) was administered without concurrent CHT. Significant regression was observed in both cranial and spinal lesions after 36 Gy (Fig. 3D-F). An MRI performed 1.5 months post-RT revealed reduced leptomeningeal enhancement and decreased size and number of cystic lesions (Fig. 3G-I). The patient’s neurological symptoms at the time of diagnosis have since completely resolved, and there is no history of seizures. Subsequently, he received carboplatin and vincristine for 1-year post-RT with the disease stabilizing.
At the latest follow-up, 27 months from diagnosis, he remains stable off therapy for 8 months, seizure-free, with no neurological deficits, developmental delay, or endocrinological abnormalities (Fig. 3J-L).
Case 3
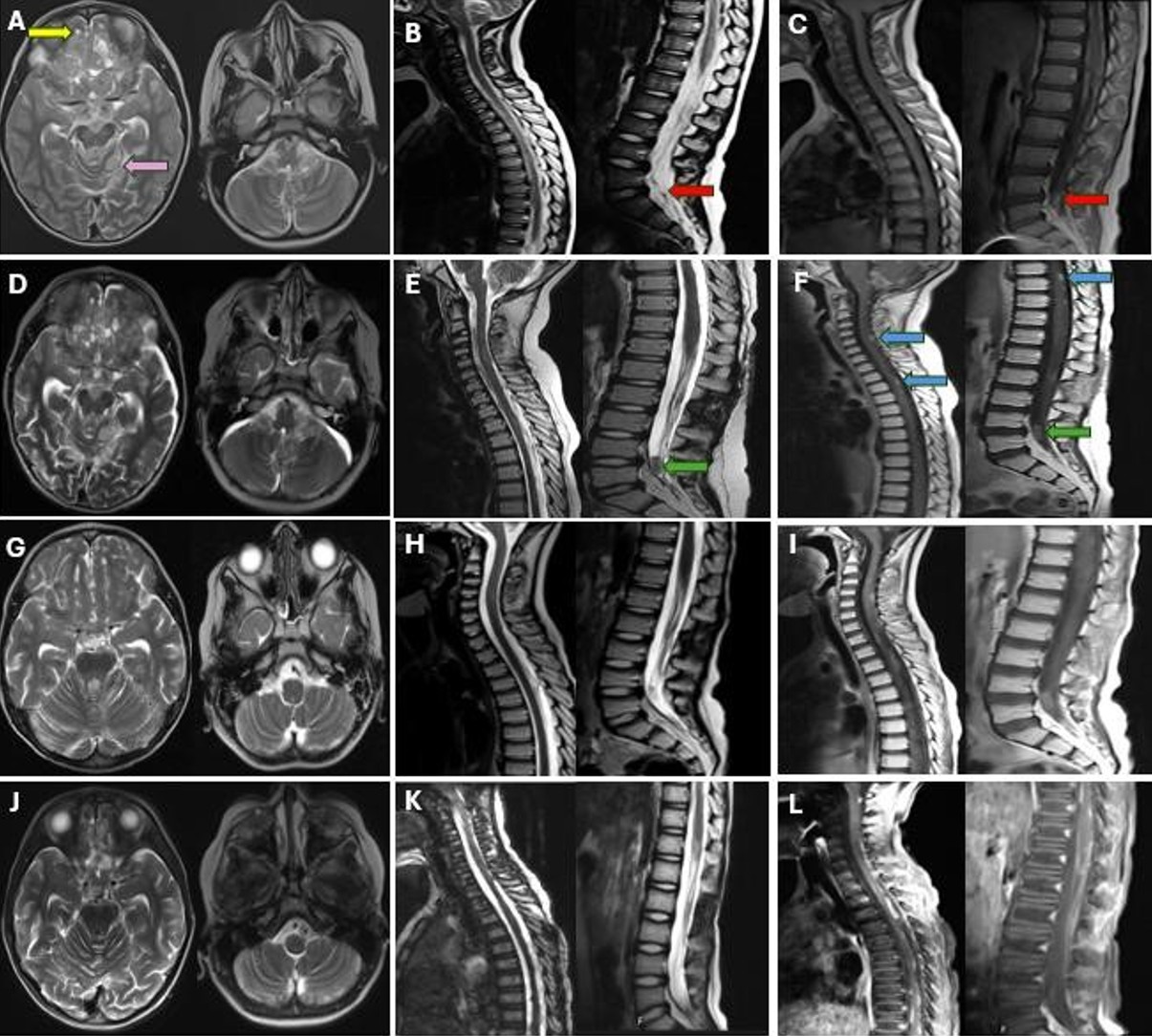
A 4-year-old boy presented with a 6-month history of seizures and headaches. Cranial MRI findings revealed leptomeningeal thickening and enhancement around the brainstem, in the basal cisterns, within the cerebellar folia, and in the sulci of both cerebral hemispheres, accompanied by occasional nodular tumor implants (Fig. 4A). Along the spinal cord, particularly at the L4-5 level, nodular tumor implants with leptomeningeal enhancement were identified (Fig. 4B, Fig. 4C).
Lumbar spinal biopsy revealed low to moderate proliferative activity, with a focal tumor area exhibiting a Ki-67/MIB-1 proliferation index of 70%, characterized by hyperchromatic nuclei and scant cytoplasm (Fig. 2C). Immunohistochemistry was consistent with DLGNT (Supplementary Table S2). CSF cytology was benign.
The patient was treated with RT consisting of 36 Gy CSI and a 54 Gy boost to the tumor bed, administered concurrently with temozolomide. Unfortunately, after the initial 36 Gy of CSI, MRI scans showed progression in both cranial and spinal regions (Fig. 4D-F). Treatment continued with a boost to the tumor bed up to 54 Gy alongside temozolomide. At 1.5 months post-RT, MRI revealed slight regression of leptomeningeal enhancement and cystic lesions (Fig. 4G-I). Despite ongoing temozolomide therapy, the patient experienced clinical and radiological progression 7 months after completing RT, particularly characterized by increased leptomeningeal thickening and enhancement in the thoracic and lumbar regions on post-contrast sequences (Fig. 4J-L). He was switched to ICE (ifosfamide, carboplatin, and etoposide) chemotherapy but died of febrile neutropenia after the first cycle. Survival after the diagnosis was 10 months.
Discussion
We report three cases of DLGNT initially treated with CSI. Two showed significant radiological response after 36 Gy without concurrent CHT, while the third progressed despite treatment and died within seven months. To our knowledge, this is the first report indicating that early radiological response to 36 Gy CSI may represent a prognostic marker in DLGNT.
The biological nature of DLGNT remains poorly understood. Consistent with several studies3,17, cytological evaluation of CSF in our series was not informative, with all CSF samples negative for tumor cells despite extensive radiological evidence of leptomeningeal involvement. DLGNTs are typically classified as low-grade gliomas (LGG) and often follow an indolent course characterized by stability or slow progression.3 In some instances, stability has been observed even without treatment.1,18 A study by Lu et al., involving 54 cases, reported a 10-year survival rate of 69%, with rates reaching 78% and 75% for patients treated with CHT alone and RT alone, respectively.19 Despite these favorable outcomes, DLGNT differs markedly from other LGGs due to its disseminated nature and unique clinical spectrum. Aggressive cases are also commonly reported, often associated with significant morbidity and long-term functional impairments.1,20 Xiao et al. noted that untreated cases had a survival range of 4 to 6 weeks, primarily due to cranial hypertension, underscoring the need for early intervention.21 Gardiman et al. reported 9 fatalities among 36 patients, with survival times ranging from 3 months to 21 years post-biopsy.1 Furthermore, a systematic review by Wiśniewski et al. reported a median overall survival (OS) of just 19 months, emphasizing the poor prognosis in certain cases.22 These findings underscore the heterogeneous nature of DLGNT, with outcomes influenced by early treatment and individual tumor characteristics.
There is currently no standardized treatment guideline for DLGNTs. Prior studies have highlighted CHT as a cornerstone in management, showing statistically significant improvements in OS.19 Common treatment protocols incorporate carboplatin, vincristine, temozolomide, cyclophosphamide, cisplatin, and/or etoposide—agents typically used in treating LGG.9,12 While CHT appears crucial, it may not suffice as monotherapy, often necessitating lifelong, multi-regimen treatment. Reports of radiological regression following CHT alone are limited, with most patients either remaining stable1,9,19,23 or showing progression.6,9,13,20,24,25 This variability underscores the need for additional or combined treatment approaches to optimize outcomes for DLGNT patients.
The role of RT in treating DLGNTs is complex, with mixed findings on its clinical and radiological efficacy. In a case series by Schniederjan et al., a patient who received CSI with temozolomide remained stable at 137 months, while another case treated with CSI alone showed progression.17 Lyle et al. reported a 14-year-old girl who, after receiving CSI with temozolomide as a first-line treatment, showed complete clinical and radiological response within six weeks, except for a residual nodule in the spine.26 Conversely, other studies suggest limited RT efficacy. Policicchio et al. indicated that the mean OS was comparable between patients treated with CHT and RT and those who were not treated (51 months vs. 53 months). Their findings also highlighted that CHT was more commonly administered in patients with better outcomes, whereas RT was slightly more prevalent in those with poorer prognoses. Among RT treated patients, 25% had poor prognostic factors, compared to only 8% in untreated patients, suggesting that RT may often be selected for patients with worse clinical characteristics.8 Supporting this, many studies employ RT primarily in cases of disease progression.12,13,27 This may be due to limited data and the perception of DLGNTs as LGGs, which has led to an expectation of limited response to RT. Furthermore, RT for DLGNT typically involves CSI, which raises concerns about potential long-term side effects, particularly for pediatric patients. In a series by Rebella et al., one case initially showed regression following CSI but subsequently developed severe leukoencephalopathy 1.5 years after treatment completion.11 - Our surviving patients, Case 1 and Case 2, exhibited a favorable response to RT and did not experience any significant side effects. Although Case 1 exhibited slight periventricular leukoencephalopathy, the patient remains asymptomatic.
Concurrent CHT with RT may be considered for aggressive cases, though risks should be individualized. Lyle et al. combined CHT and CSI based on rapid, sustained responses.26 In contrast, our Case 3 progressed despite concurrent temozolomide with CSI, highlighting the need for biomarkers to identify aggressive subtypes.
To better identify patients at risk for aggressive disease, preliminary data suggest prioritizing molecular features over traditional clinical approaches. A review by Policicchio et al., covering both adult and pediatric populations, found that Ki-67/MIB-1 levels in pediatric studies typically ranged from 1% to 30%. Karlowee et al. reported a Ki-67/MIB-1 index of 40%, while Swetye et al. observed focal Ki-67/MIB-1 levels reaching 53% in this review. A cutoff of 5% has also been suggested. An average OS of 46 months was observed for patients with Ki-67/MIB-1 levels between 0% and 5%. In contrast, those with Ki-67/MIB-1 levels exceeding 5% experienced a mortality rate of 36% (7 out of 19 patients) and an average OS of only 8.8 months.8 Additionally, Wiśniewski et al. identified a Ki-67/MIB-1 index greater than 7% as the most significant prognostic factor for OS in patients with DLGNT.22 Rodriguez et al. further identified that a Ki-67/MIB-1 index over 4%, mitotic activity exceeding 4 mitoses per high-power field, or the presence of glomeruloid vasculature correlated with poor survival, collectively suggesting anaplasia when any of these criteria are present.2 Schniederjan et al. described aggressive histologic features, including necrosis, mitotic figures, nuclear pleomorphism, and microvascular proliferation, as indicators of anaplastic behavior.17 Another study also noted polar spongioblastoma patterns and tumor invasion into brain parenchyma as indicators of anaplasia, though identified only in postmortem tissue.28 According to Swetye et al.’s case, with a Ki-67/MIB-1 proliferative index of 53% and increased mitotic activity, aligned with the anaplastic features defined by Rodriguez et al. and demonstrated rapid progression within 6 months.24 It can be inferred from this literature that a reported Ki-67/MIB-1 level of 4% does not reliably indicate anaplasia, particularly when compared to studies that use differing cutoff values. Also, it remains uncertain whether these anaplastic features are linked to more aggressive clinical behavior. In our study, - Case 3 did not exhibit glomeruloid vasculature, high mitotic activity, necrosis, or microvascular proliferation, though a focal Ki-67/MIB-1 index of 70% —the highest reported in the literature— might suggest anaplasia. The patient showed only a transient response after CSI, with progression by 7 months and eventual death. This suggests a link between anaplastic features, lack of early radiological response, and poor prognosis.
Currently, additional immunohistochemical and molecular markers for DLGNTs are still under investigation. While MAP-2 has been linked to poor survival, our Cases 1 and 2 showed favorable outcomes, highlighting variability among patients.22 The role of molecular alterations, such as BRAF mutations, in prognosis remains under examination2; notably, our Case 1 and Case 3 did not demonstrate these mutations. Deng et al. proposed two methylation subtypes (DLGNT-MC-1 with 1p/19q codeletion and DLGNT-MC-2 with 1p deletion/1q gain), suggesting a grading system resembling CNS WHO grades II and III.5 However, data on these classifications remain limited, and our inability to perform such analyses is a key limitation, emphasizing the need for further research to refine prognosis and treatment strategies in DLGNT.
Our study emphasizes the importance of CSI in treating DLGNTs. Although the small sample size of three cases limits statistical power, this study serves as a valuable reference for future research in this rare tumor entity. We believe that an early favorable response to RT may be associated with a better prognosis. We emphasize the need for future research to establish more reliable prognostic markers and to refine therapeutic strategies.
Ethical approval
The study was approved by Ege University Medical Research Ethics Committee (date: 22.08.2024, number: 24-8T/52).
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Gardiman MP, Fassan M, Orvieto E, et al. Diffuse leptomeningeal glioneuronal tumors: a new entity? Brain Pathol 2010; 20: 361-366. https://doi.org/10.1111/j.1750-3639.2009.00285.x
- Rodriguez FJ, Schniederjan MJ, Nicolaides T, Tihan T, Burger PC, Perry A. High rate of concurrent BRAF-KIAA1549 gene fusion and 1p deletion in Disseminated Oligodendroglioma-like Leptomeningeal Neoplasms (DOLN). Acta Neuropathol 2015; 129: 609-610. https://doi.org/10.1007/s00401-015-1400-9
- Rodriguez FJ, Perry A, Rosenblum MK, et al. Disseminated oligodendroglial-like leptomeningeal tumor of childhood: a distinctive clinicopathologic entity. Acta Neuropathol 2012; 124: 627-641. https://doi.org/10.1007/s00401-012-1037-x
- Louis DN, Perry A, Wesseling P, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol 2021; 23: 1231-1251. https://doi.org/10.1093/neuonc/noab106
- Deng MY, Sill M, Chiang J, et al. Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol 2018; 136: 239-253. https://doi.org/10.1007/s00401-018-1865-4
- Ozen A, Tanrikulu B, Danyeli AE, Ozek MM. Pediatric diffuse Leptomeningeal Glioneuronal tumors: diagnosis, follow-up, and treatment options. Turk Neurosurg 2024; 34: 441-447. https://doi.org/10.5137/1019-5149.JTN.43742-23.2
- Abongwa C, Cotter J, Tamrazi B, Dhall G, Davidson T, Margol A. Primary diffuse leptomeningeal glioneuronal tumors of the central nervous system: report of three cases and review of literature. Pediatr Hematol Oncol 2020; 37: 248-258. https://doi.org/10.1080/08880018.2019.1711270
- Policicchio D, Boccaletti R, Cuccu AS, et al. Atypical and aggressive diffuse leptomeningeal glioneuronal tumor in a young adult: a case report and review of the literature. Surg Neurol Int 2022; 13: 214. https://doi.org/10.25259/SNI_1255_2021
- Aguilera D, Castellino RC, Janss A, et al. Clinical responses of patients with diffuse leptomeningeal glioneuronal tumors to chemotherapy. Childs Nerv Syst 2018; 34: 329-334. https://doi.org/10.1007/s00381-017-3584-x
- Bajin IY, Levine A, Dewan MC, et al. Understanding diffuse leptomeningeal glioneuronal tumors. Childs Nerv Syst 2024; 40: 2359-2366. https://doi.org/10.1007/s00381-024-06432-6
- Rebella G, Milanaccio C, Morana G, et al. Calcifications in diffuse leptomeningeal glioneuronal tumors: a case series. Quant Imaging Med Surg 2022; 12: 2985-2994. https://doi.org/10.21037/qims-21-741
- Dodgshun AJ, SantaCruz N, Hwang J, et al. Disseminated glioneuronal tumors occurring in childhood: treatment outcomes and BRAF alterations including V600E mutation. J Neurooncol 2016; 128: 293-302. https://doi.org/10.1007/s11060-016-2109-x
- Preuss M, Christiansen H, Merkenschlager A, et al. Disseminated oligodendroglial-like leptomeningeal tumors: preliminary diagnostic and therapeutic results for a novel tumor entity. J Neurooncol 2015; 124: 65-74. https://doi.org/10.1007/s11060-015-1735-z
- Xu H, Chen F, Zhu H, Luo L, Zhang R. Diffuse leptomeningeal glioneuronal tumor in a Chinese adult: a novel case report and review of literature. Acta Neurol Belg 2020; 120: 247-256. https://doi.org/10.1007/s13760-019-01262-9
- Al-Ghanem R, Luque Barona R, Godoy-Hurtado A, Galicia Bulnes JM, El-Rubaidi O. Diffuse leptomeningeal glioneuronal tumor: a review of diagnosis and management with an illustrative case. Neurocirugia 2022; 33: 389-393. https://doi.org/10.1016/j.neucie.2022.02.006
- Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol 2010; 28: 1963-1972. https://doi.org/10.1200/JCO.2009.26.3541
- Schniederjan MJ, Alghamdi S, Castellano-Sanchez A, et al. Diffuse leptomeningeal neuroepithelial tumor: 9 pediatric cases with chromosome 1p/19q deletion status and IDH1 (R132H) immunohistochemistry. Am J Surg Pathol 2013; 37: 763-771. https://doi.org/10.1097/PAS.0b013e31827bf4cc
- Patankar AP, Vaghela P, Nasit J, Gohil R. Diffuse leptomeningeal glioneuronal tumor: a rare case report with review of literature. Asian J Neurosurg 2022; 17: 532-535. https://doi.org/10.1055/s-0042-1756637
- Lu VM, Di L, Gernsback J, et al. Contemporary outcomes of diffuse leptomeningeal glioneuronal tumor in pediatric patients: a case series and literature review. Clin Neurol Neurosurg 2022; 218: 107265. https://doi.org/10.1016/j.clineuro.2022.107265
- Cho HJ, Myung JK, Kim H, et al. Primary diffuse leptomeningeal glioneuronal tumors. Brain Tumor Pathol 2015; 32: 49-55. https://doi.org/10.1007/s10014-014-0187-z
- Xiao J, Gao L, Zhang M, et al. Clinical features of diffuse Leptomeningeal Glioneuronal Tumor with rapid blindness misdiagnosed as NMOSD and literature review. SN Compr Clin Med 2019; 1: 434-441. https://doi.org/10.1007/s42399-019-00058-5
- Wiśniewski K, Brandel MG, Gonda DD, Crawford JR, Levy ML. Prognostic factors in diffuse leptomeningeal glioneuronal tumor (DLGNT): a systematic review. Childs Nerv Syst 2022; 38: 1663-1673. https://doi.org/10.1007/s00381-022-05600-w
- Manoharan N, Ajuyah P, Senapati A, et al. Diffuse Leptomeningeal Glioneuronal Tumour (DLGNT) in children: the emerging role of genomic analysis. Acta Neuropathol Commun 2021; 9: 147. https://doi.org/10.1186/s40478-021-01248-w
- Schwetye KE, Kansagra AP, McEachern J, Schmidt RE, Gauvain K, Dahiya S. Unusual high-grade features in pediatric diffuse leptomeningeal glioneuronal tumor: comparison with a typical low-grade example. Hum Pathol 2017; 70: 105-112. https://doi.org/10.1016/j.humpath.2017.06.004
- Garibotto F, Pavanello M, Milanaccio C, Gaggero G, Fiaschi P. Management of hydrocephalus related to diffuse leptomeningeal glioneuronal tumour: a multifaceted condition. Childs Nerv Syst 2021; 37: 1039-1040. https://doi.org/10.1007/s00381-020-04867-1
- Lyle MR, Dolia JN, Fratkin J, Nichols TA, Herrington BL. Newly identified characteristics and suggestions for diagnosis and treatment of diffuse leptomeningeal glioneuronal/neuroepithelial tumors: a case report and review of the literature. Child Neurol Open 2015; 2: 2329048X14567531. https://doi.org/10.1177/2329048X14567531
- Karlowee V, Kolakshyapati M, Amatya VJ, et al. Diffuse Leptomeningeal Glioneuronal Tumor (DLGNT) mimicking Whipple’s disease: a case report and literature review. Childs Nerv Syst 2017; 33: 1411-1414. https://doi.org/10.1007/s00381-017-3405-2
- Yamasaki T, Sakai N, Shinmura K, et al. Anaplastic changes of diffuse leptomeningeal glioneuronal tumor with polar spongioblastoma pattern. Brain Tumor Pathol 2018; 35: 209-216. https://doi.org/10.1007/s10014-018-0326-z
Copyright and license
Copyright © 2025 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









