
Abstract
Background. Autoimmune manifestations are increasingly recognized as late complications following hematopoietic stem cell transplantation (HSCT), particularly in association with chronic graft-versus-host disease (cGvHD). However, the occurrence of systemic lupus erythematosus (SLE)-like features remains extremely rare, especially in pediatric patients. Understanding the mechanisms underlying such manifestations, including the role of mixed chimerism, is important for early recognition and management.
Case Presentation. We report the case of a girl with thalassemia major who underwent HSCT from a fully matched unrelated donor at the age of six years. Her early post-transplant course was complicated by gastrointestinal cGvHD, followed by autoimmune hemolytic anemia and arthritis, which responded to corticosteroids and methotrexate. Four years post-HSCT, she developed a lupus-like syndrome characterized by malar rash, serositis, cytopenias, arthritis, high-titer antinuclear antibodies (ANA), elevated anti-double-stranded DNA (anti-dsDNA) antibodies, and low complement levels, fulfilling the American College of Rheumatology classification criteria for SLE. At the time of symptom onset, mixed chimerism was documented, with a notable proportion of recipient-derived lymphocytes. Treatment with mycophenolate mofetil and hydroxychloroquine led to rapid clinical and laboratory improvement.
Conclusion. This case illustrates the evolving nature of post-transplant immune dysregulation and suggests that declining donor chimerism may contribute to the reactivation of autoreactive lymphocytes, leading to atypical autoimmune manifestations. In pediatric patients presenting with unusual post-transplant symptoms, careful clinical assessment and immune monitoring may aid in timely diagnosis. Individualized immunosuppressive therapy can facilitate symptom control and support favorable long-term outcomes.
Keywords: autoimmune diseases, graft vs host disease, hematopoietic stem cell transplantation, systemic lupus erythematosus
Introduction
Autoimmune complications are increasingly recognized as a late effect of hematopoietic stem cell transplantation (HSCT), and they can arise following transplantation for both malignant and non-malignant hematologic conditions. The most commonly encountered autoimmune complications are immune-mediated cytopenias, including autoimmune hemolytic anemia, immune thrombocytopenia, and neutropenia.1 These manifestations are thought to arise from a breakdown of central and peripheral immune tolerance mechanisms. While dysfunction of regulatory T cells (Tregs), which normally suppress autoreactive lymphocytes, plays a central role, additional mechanisms have also been implicated, including impaired thymic reconstitution, dysregulated B-cell homeostasis with autoantibody production, aberrant cytokine signaling, and delayed immune reconstitution following HSCT.2-4 Disruption of these tightly regulated processes may ultimately lead to unrestrained autoreactivity and the development of autoimmune disease.
Chronic graft-versus-host disease (cGvHD) remains a significant and often debilitating complication of allogeneic HSCT, with incidence rates reported between 30% and 70%, depending on various transplant-related factors.5 The disease is marked by a wide spectrum of clinical presentations and may closely resemble classical autoimmune disorders, including systemic sclerosis, primary biliary cholangitis, and Sjögren’s syndrome.6,7
Chimerism represents a key immunological factor implicated in the development of post-transplant autoimmunity. It refers to the coexistence of donor- and recipient-derived hematopoietic cells following HSCT and is typically classified as full donor chimerism or mixed chimerism. In the setting of mixed chimerism, the persistence of recipient immune cells alongside donor cells may contribute to immune dysregulation through impaired immune tolerance, altered antigen presentation, and ongoing immune activation.8 Accumulating evidence indicates that mixed chimerism may predispose patients to autoimmune phenomena after HSCT, underscoring the importance of regular chimerism monitoring during long-term follow-up.
Although autoimmune features are frequently observed in cGvHD, lupus-like presentations remain exceedingly rare and are only sporadically reported. In this report, we present an unusual case of cGvHD that manifested with clinical and serological features strongly suggestive of systemic lupus erythematosus (SLE).
Case Presentation
A female infant was diagnosed with thalassemia major at six months of age during the evaluation of anemia. From early infancy, she required regular red blood cell transfusions. Due to the chronic transfusion burden, her serum ferritin level was measured at 1082 ng/mL (normal range: 10-290 ng/mL) prior to transplantation to assess iron overload. At six years of age, she underwent a fully HLA-matched (10/10) allogeneic HSCT from a matched unrelated donor (MUD). The myeloablative conditioning regimen, which included treosulfan, fludarabine, cyclophosphamide, thiotepa, and antithymocyte globulin (ATG), was followed by GvHD prophylaxis with cyclosporine and low-dose methotrexate. Early post-transplant chimerism analysis revealed 99% donor cell engraftment. Following transplantation, a post-HSCT ferritin level was measured at 678 ng/mL.
On day +42 post-HSCT, the patient presented with fever, diarrhea, and vomiting, prompting a colonoscopy which revealed diffuse mucosal edema and aphthous ulcerations. The histopathological analysis was consistent with grade 3 gastrointestinal GvHD, showing prominent apoptosis, crypt loss, and crypt abscesses. Treatment with methylprednisolone (2 mg/kg/day) resulted in rapid clinical improvement. Full donor chimerism was maintained, and corticosteroids were tapered and discontinued by six months post-transplantation. Around one year after transplantation, the patient developed autoimmune hemolytic anemia, evidenced by a hemoglobin level of 7 g/dL and a positive direct Coombs test. There was no associated GvHD or infectious trigger. Treatment with oral corticosteroids led to prompt clinical improvement and was continued for four months before successful discontinuation without relapse, while cyclosporine was ceased at 17 months post-HSCT.
Approximately three years after HSCT, the patient presented to the pediatric rheumatology clinic with an eight-month history of bilateral knee and wrist pain, associated with morning stiffness lasting approximately one hour. At that time, she maintained mixed chimerism (51%), remained transfusion-independent, and had no clinical evidence of ongoing GvHD. On physical examination, arthritis involving bilateral wrists, knees, and temporomandibular joints was observed. Laboratory testing revealed anemia (hemoglobin: 8.2 g/dL) and a positive direct Coombs test, while ANA, anti-dsDNA, and rheumatoid factor were negative. Complement levels (C3, C4) were within normal limits. The clinical presentation was suggestive of juvenile idiopathic arthritis (JIA) and autoimmune hemolytic anemia, prompting initiation of therapy with methotrexate and methylprednisolone, which led to clinical improvement. However, mild to moderate anemia persisted despite treatment. Corticosteroids were gradually tapered and successfully discontinued by the third month.
At four years post-transplant, and one year following the diagnosis of JIA, the patient presented with malar rash, fatigue, hair loss, and recurrence of joint symptoms, despite ongoing methotrexate therapy. Physical examination confirmed the presence of malar rash and arthritis of the wrists. Laboratory tests revealed cytopenias: hemoglobin 8 g/dL, neutropenia (550/μL), lymphopenia (1,300/μL), and a normal platelet count. Bone marrow biopsy demonstrated preserved trilineage hematopoiesis. Screening for viral infections was negative, and there was no proteinuria. Erythrocyte sedimentation rate (ESR) was elevated at 96 mm/h. Direct Coombs test remained positive. Autoimmune workup revealed high-titer ANA (1:3200, homogeneous pattern), elevated anti-dsDNA (171 IU/L (normal range: <100 IU/ml). Complement analysis showed a decreased C4 level (<10.06 mg/dL; normal: 12–36 mg/dL), while C3 was within the normal range (89.6 mg/dL; normal: 85–160 mg/dL). Liver function tests were within normal limits (AST: 28 U/L [normal: 5–40], ALT: 32 U/L [normal: 10–40]), and kidney function was normal (serum creatinine: 0.44 mg/dL, urea: 19 mg/dL [normal: 19–49]). Urinalysis was unremarkable, and the urinary protein-to-creatinine ratio was 0.2, indicating no significant proteinuria. Extended immunological evaluation revealed normal serum immunoglobulin levels (IgG, IgA, and IgM), normal lymphocyte subsets, and negative extended autoimmune serology, including extractable nuclear antigen (ENA) panel and antiphospholipid antibodies. Transthoracic echocardiography revealed pericardial effusion. At the time of lupus-like disease onset, chimerism analysis demonstrated 25% recipient-derived T cells and 51% recipient-derived B cells.
This constellation of findings fulfilled the classification criteria for SLE according to the American College of Rheumatology (ACR), the European League Against Rheumatism/American College of Rheumatology (EULAR/ACR), and the Systemic Lupus International Collaborating Clinics (SLICC), including: serositis, arthritis, malar rash, Coombs-positive hemolytic anemia, leukopenia, positive ANA, and anti-dsDNA antibodies. The diagnosis was therefore revised to SLE-like disease following HSCT. Treatment with mycophenolate mofetil and hydroxychloroquine was initiated, resulting in complete clinical and laboratory remission within the first month. Fig. 1 outlines the timeline of major clinical events and treatments. She has been followed up for six months with no signs of disease activation. At the most recent visit, complement levels were within the normal range, ANA was positive at a titer of 1:160, and anti-dsDNA was negative.
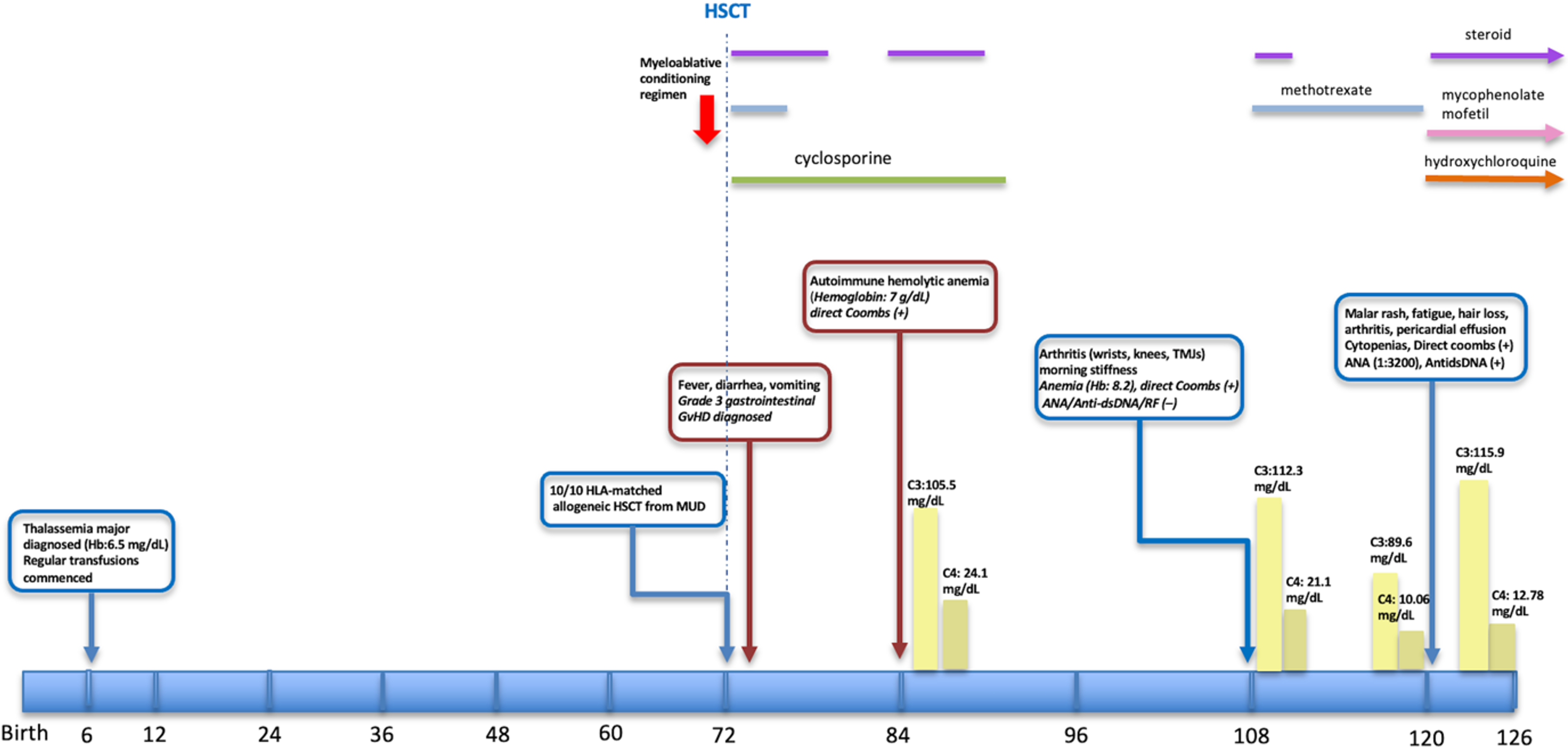
ANA, antinuclear antibody; anti-dsDNA, anti–double-stranded DNA antibody; GvHD, graft-versus-host disease; Hb, hemoglobin; HLA, human leukocyte antigen; HSCT, hematopoietic stem cell transplantation; MUD, matched unrelated donor; RF, rheumatoid factor; TMJ, temporomandibular joint
Written informed consent was obtained from the patient’s parents for the publication of this case report.
Discussion
A growing range of autoimmune phenomena has been recognized as late complications of allogeneic HSCT. Although post-transplant immune dysregulation may resemble several autoimmune diseases, the manifestation of SLE-like features that meet established classification criteria is exceedingly uncommon.
The present case demonstrates a complex and evolving autoimmune course in a pediatric patient who developed a lupus-like syndrome with overlapping features of chronic GvHD and de novo autoimmunity four years after HSCT. This observation raises important questions regarding the immunopathogenesis underlying post-HSCT autoimmunity and how it differs from classical cGvHD.
One proposed mechanism involves reconstitution of a dysregulated immune system, in which impaired central and peripheral tolerance allows autoreactive T and B lymphocytes to survive and expand.9 Recovery of Tregs following HSCT may be incomplete, or they may be functionally impaired, particularly in the context of prior GvHD and prolonged immunosuppressive therapy. Furthermore, as observed in our patient, mixed chimerism may indicate incomplete immune replacement, with persistence of autoreactive recipient-derived lymphocytes potentially contributing to autoimmunity through impaired tolerance, altered antigen presentation, and ongoing immune activation.10 The chronological progression from initial gastrointestinal GvHD to the subsequent development of juvenile idiopathic arthritis and, eventually, a lupus-like phenotype raises the possibility that post-transplant immune dysregulation potentially driven by declining donor chimerism may underlie this evolving autoimmune spectrum, allowing reactivation of autoreactive immune cells and breakdown of tolerance. Chimerism monitoring may therefore serve not only as a marker of graft stability but also as a clinically relevant immunologic indicator in patients who develop late autoimmune manifestations. In particular, declining donor chimerism or lineage specific recipient predominance (e.g., T- or B-cell fractions) may reflect incomplete immune replacement and impaired tolerance reestablishment.11 While evidence remains limited, our case supports the practice of regular, lineage specific chimerism assessment in long term follow-up, especially when new autoimmune symptoms or atypical inflammatory findings emerge.
Although the prevalence of cGvHD is lower in pediatric patients than in adults (approximately 20–50% vs. 60–70% in most series), it remains a clinically important complication in children, particularly as the use of peripheral blood stem cells and unrelated donors becomes more widespread.12,13 Acute GvHD typically involves the skin, liver, and gastrointestinal tract, whereas chronic GvHD has a broader spectrum and may affect almost any organ. The updated 2020 National Institutes of Health (NIH) diagnostic criteria expanded the definition of cGvHD to include atypical features such as serositis, nephrotic syndrome, and cytopenias; these features were previously excluded under the classical definitions.14 Autoimmune phenomena, including cytopenias and arthritis, are now increasingly recognized in the context of cGvHD.
Systemic lupus erythematosus–like after HSCT has been reported only sporadically, mostly as isolated case reports. Stylianou et al. described a lupus-like syndrome with renal involvement after allogeneic HSCT in the context of cGvHD, highlighting the clinical resemblance yet potential immunologic distinctiveness of post-transplant autoimmunity.15 A pediatric case of SLE occurring years after unrelated cord blood transplantation has also been reported, underscoring that lupus phenotypes may emerge late in the post-transplant period.16 These reports, together with our case, suggest that lupus-like phenotypes can occur across age groups and transplant settings, often with complex overlaps between alloimmune injury (cGvHD) and de novo autoimmunity.
There is a need for treatment guidelines specific to atypical forms of cGvHD, particularly those with autoimmune overlap, as therapeutic approaches often differ from classic cGvHD and remain limited. This need was emphasized in the 2025 NIH Chronic Graft-versus-Host Disease Consensus Conference update.17 Chronic GvHD is typically managed with corticosteroids, either as monotherapy or in combination with other immunosuppressive agents (tacrolimus, methotrexate, cyclosporine, mycophenolate mofetil), achieving a complete remission rate of 63.5%.18 While literature remains limited, a previously reported case of lupus-like syndrome presenting with immune complex-mediated diffuse proliferative glomerulonephritis following HSCT responded well to treatment with corticosteroids, cyclophosphamide, and mycophenolate mofetil.19 Complete clinical and laboratory remission was achieved with the combination of corticosteroids and mycophenolate mofetil in our patient.
Beyond the post-transplant setting, chimerism/microchimerism has long been investigated as a potential contributor to autoimmunity. Microchimerism refers to the presence of a small population of genetically distinct cells (most commonly maternal–fetal) within an individual and has been described in association with autoimmune diseases, including SLE.20 Studies evaluating maternal microchimerism in SLE have reported variable results, reflecting heterogeneity in detection methods and clinical phenotypes.21 In juvenile inflammatory myopathies, including juvenile dermatomyositis, microchimeric cells have also been investigated; available data do not consistently support a direct pathogenic role, but they reinforce the concept that chimerism related immune tolerance mechanisms may intersect with pediatric autoimmunity.22 Although microchimerism differs biologically from post-HSCT mixed hematopoietic chimerism, both phenomena underscore the broader principle that the coexistence of genetically distinct immune cell populations may influence immune tolerance and autoimmunity.
In conclusion, this case underscores the multifaceted and evolving nature of immune dysregulation in pediatric recipients of hematopoietic stem cell transplantation and highlights the critical importance of long-term immunologic surveillance, including serial chimerism analysis. Although the patient is currently in clinical remission, this status should be interpreted with caution, as the duration of follow-up following the onset of lupus-like disease remains limited. Given the dynamic immune reconstitution after HSCT and the presence of mixed chimerism, the risk of disease relapse or progression toward a more systemic autoimmune phenotype cannot be excluded. Therefore, prolonged, and comprehensive follow-up is warranted to monitor for potential reactivation of autoimmune manifestations or the emergence of additional systemic features over time.
Ethical approval
Written informed consent was obtained from the patient and her legal guardians for publication of this case report and accompanying images. Ethical committee approval was not obtained, as it was not required for a single case report according to our institutional policy.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Hartert A, Willenbacher W, Günzelmann S, et al. Successful treatment of thrombocytopenia and hemolytic anemia with IvIG in a patient with lupus-like syndrome after mismatched related PBSCT. Bone Marrow Transplant 2001; 27: 337-340. https://doi.org/10.1038/sj.bmt.1702774
- Marleau AM, Sarvetnick N. T cell homeostasis in tolerance and immunity. J Leukoc Biol 2005; 78: 575-584. https://doi.org/10.1189/jlb.0105050
- Daikeler T, Labopin M, Di Gioia M, et al. Secondary autoimmune diseases occurring after HSCT for an autoimmune disease: a retrospective study of the EBMT Autoimmune Disease Working Party. Blood 2011; 118: 1693-1698. https://doi.org/10.1182/blood-2011-02-336156
- Storek J, Geddes M, Khan F, et al. Reconstitution of the immune system after hematopoietic stem cell transplantation in humans. Semin Immunopathol 2008; 30: 425-437. https://doi.org/10.1007/s00281-008-0132-5
- Lee SJ, Flowers MED. Recognizing and managing chronic graft-versus-host disease. Hematology Am Soc Hematol Educ Program 2008; 1: 134-141. https://doi.org/10.1182/asheducation-2008.1.134
- Mavragani CP, Moutsopoulos HM. The geoepidemiology of Sjögren’s syndrome. Autoimmun Rev 2010; 9: A305-A310. https://doi.org/10.1016/j.autrev.2009.11.004
- Tyndall A, Gratwohl A. Blood and marrow stem cell transplants in autoimmune disease. A consensus report written on behalf of the European League Against Rheumatism (EULAR) and the European Group for Blood and Marrow Transplantation (EBMT). Br J Rheumatol 1997; 36: 390-392. https://doi.org/10.1093/rheumatology/36.3.390
- Daikeler T, Labopin M, Ruggeri A, et al. New autoimmune diseases after cord blood transplantation: a retrospective study of EUROCORD and the Autoimmune Disease Working Party of the European Group for Blood and Marrow Transplantation. Blood 2013; 121: 1059-1064. https://doi.org/10.1182/blood-2012-07-445965
- Moon JH, Lee SJ, Kim JG, et al. Clinical significance of autoantibody expression in allogeneic stem-cell recipients. Transplantation 2009; 88: 242-250. https://doi.org/10.1097/TP.0b013e3181ac6885
- Buxbaum NP, Pavletic SZ. Autoimmunity following allogeneic hematopoietic stem cell transplantation. Front Immunol 2020; 11: 2017. https://doi.org/10.3389/fimmu.2020.02017
- Park M, Koh KN, Seo JJ, Im HJ. Clinical implications of chimerism after allogeneic hematopoietic stem cell transplantation in children with non-malignant diseases. Korean J Hematol 2011; 46: 258-264. https://doi.org/10.5045/kjh.2011.46.4.258
- Baird K, Cooke K, Schultz KR. Chronic graft-versus-host disease (GVHD) in children. Pediatr Clin North Am 2010; 57: 297-322. https://doi.org/10.1016/j.pcl.2009.11.003
- Ochs LA, Miller WJ, Filipovich AH, et al. Predictive factors for chronic graft-versus-host disease after histocompatible sibling donor bone marrow transplantation. Bone Marrow Transplant 1994; 13: 455-460.
- Kitko CL, Pidala J, Schoemans HM, et al. National Institutes of Health Consensus Development Project on criteria for clinical trials in chronic graft-versus-host disease: iia. The 2020 clinical implementation and early diagnosis working group report. Transplant Cell Ther 2021; 27: 545-557. https://doi.org/10.1016/j.jtct.2021.03.033
- Stylianou K, Stratakis S, Mavroeidi V, et al. Membranous nephropathy and lupus-like syndrome after hematopoietic cell transplantation: a case report. J Med Case Rep 2010; 4: 303. https://doi.org/10.1186/1752-1947-4-303
- Nagasawa M, Aoki Y. A pediatric case of systemic lupus erythematosus developed 10 years after cord blood transplantation for juvenile myelomonocytic leukemia. Case Rep Transplant 2012; 619126. https://doi.org/10.1155/2012/619126
- Lee SJ, Williams KM, Sarantopoulos S, et al. NIH chronic graft-versus-host disease consensus conference 2025 update. Transplant Cell Ther 2025; 31: 678.e1-678.e16. https://doi.org/10.1016/j.jtct.2025.05.016
- McManigle W, Youssef A, Sarantopoulos S. B cells in chronic graft-versus-host disease. Hum Immunol 2019; 80: 393-399. https://doi.org/10.1016/j.humimm.2019.03.003
- Yang J, Zhang L, Sha W, Liu S, Shen L. Lupus-like manifestations after allogenic hematopoietic stem cell transplantation: a rare case of chronic graft-versus-host disease. J Nephrol 2025; 38: 1239-1244. https://doi.org/10.1007/s40620-024-01988-7
- Régnier S, Aractingi S. Microchimerism and autoimmune diseases. Eur J Intern Med 2002; 13: 365. https://doi.org/10.1016/s0953-6205(02)00098-5
- Kanold AMJ, Svenungsson E, Gunnarsson I, et al. A research study of the association between maternal microchimerism and systemic lupus erythematosus in adults: a comparison between patients and healthy controls based on single-nucleotide polymorphism using quantitative real-time PCR. PLoS One 2013; 8: e74534. https://doi.org/10.1371/journal.pone.0074534
- Artlett CM, Sassi-Gaha S, Ramos RC, Miller FW, Rider LG. Chimeric cells of maternal origin do not appear to be pathogenic in the juvenile idiopathic inflammatory myopathies or muscular dystrophy. Arthritis Res Ther 2015; 17: 238. https://doi.org/10.1186/s13075-015-0732-0
Copyright and license
Copyright © 2026 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









