
Abstract
Background. The 16p11.2 deletion is one of the most frequent recurrent copy number variations associated with a broad neurodevelopmental and phenotypic spectrum. Despite its relatively well-characterized genomic region, clinical expressivity remains highly variable, posing challenges for diagnosis and management.
Methods. We conducted a retrospective single-centre study of 25 individuals with molecularly confirmed 16p11.2 deletions, including 13 males (52%), 12 females (48%), and 7 familial (28%). Both de novo and inherited cases were included. The main testing method was chromosomal microarray, although karyotyping and additional tests such as sequencing and trinucleotide repeat testing were also utilized. Comprehensive clinical data were collected from medical records, including neurodevelopmental, neuropsychiatric, metabolic, skeletal, and systemic features.
Results. The majority of the cases had the typical ~600 kilobase deletion while two had distal ~220kb deletion. One patient was found to have a double genetic diagnosis. Developmental delay was almost universal in the probands, with expressive language significantly more impaired than receptive language abilities. Intellectual disability / learning difficulties and language problems were observed in 18/25 (72%) cases. Around half of the probands showed obesity and related hyperphagia. Autism spectrum disorder, attention deficit hyperactivity disorder, stereotypic movements, and aggressive behaviour were frequently reported. Epilepsy was present in thirteen patients (52%), with electroencephalographic abnormalities supporting generalized or focal epileptiform activity. Dysmorphic facial features and skeletal anomalies such as pes equinovarus, syndactyly, and scoliosis were variably present. Brain magnetic resonance imaging revealed abnormalities in several patients, including hypoplasia of the corpus callosum and intracranial hypertension. Additional systemic findings included hepatic steatosis, constipation, and ophthalmologic anomalies. Parental testing revealed asymptomatic or mildly affected carriers in multiple cases.
Conclusion. Our findings emphasize the broad and heterogeneous clinical spectrum of 16p11.2 deletions in a Turkish cohort. Early recognition, multidisciplinary evaluation, and family-based genetic counselling are essential for timely diagnosis and optimal care of affected individuals.
Keywords: 16p11.2 deletion, phenotypic variability, genotype-phenotype correlation, Turkish cohort
Introduction
The 16p11.2 region of the human genome is enriched with low copy repeats, making it prone to misalignment during meiotic recombination. This susceptibility often results in non-allelic homologous recombination, leading to recurrent copy number variations (CNVs) at specific breakpoints. Among these, deletions and duplications of 16p11.2 are recognized as among the most frequently observed pathogenic CNVs associated with neurodevelopmental disorders.1 Despite notable clinical variability, affected individuals often share overlapping features such as developmental delay (DD), intellectual disability (ID), neuropsychiatric symptoms, obesity or underweight, congenital anomalies, and epilepsy.2
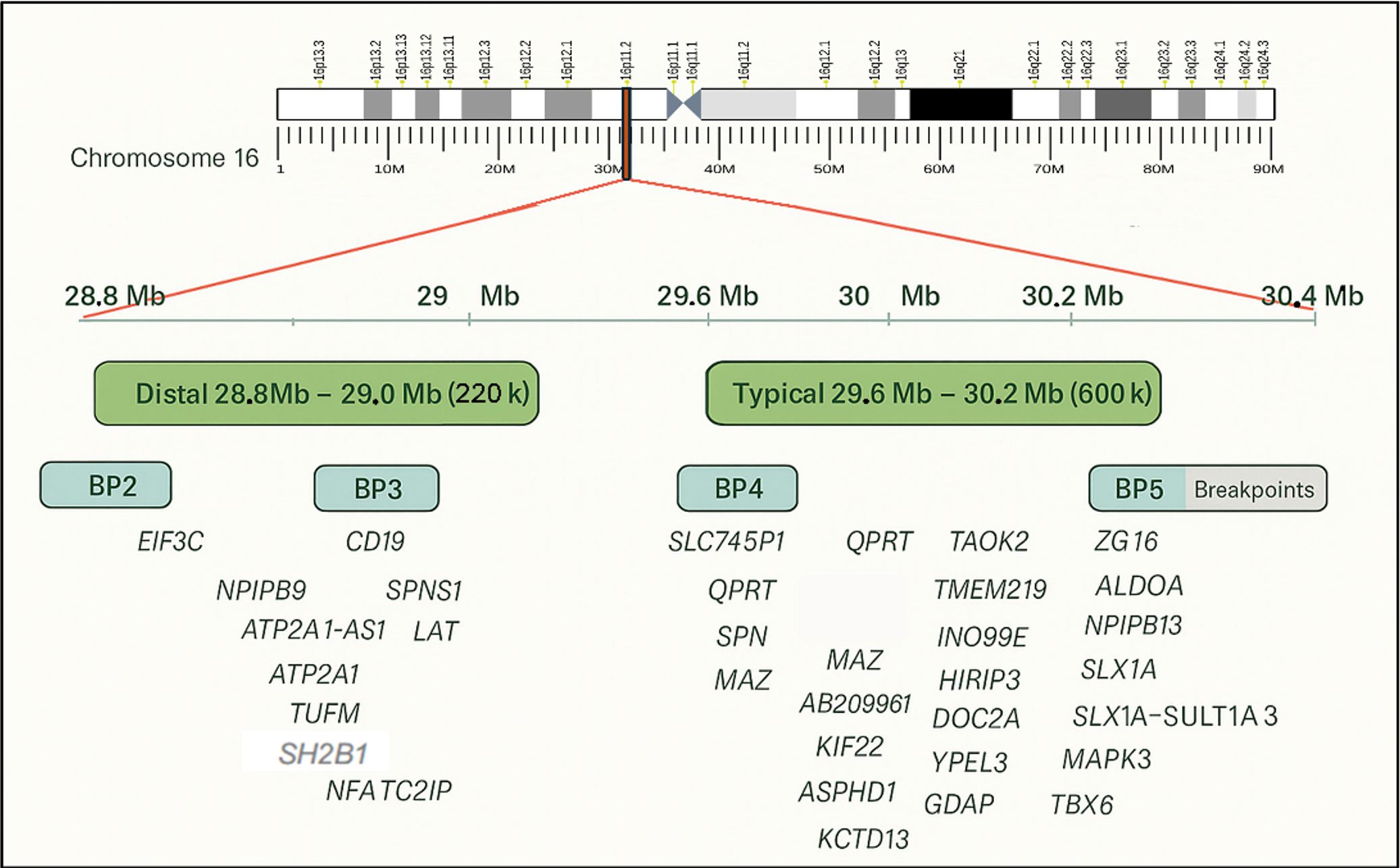
The well-characterized “typical” 16p11.2 deletion spans the BP4–BP5 region (OMIM #611913), covering approximately 600 kilobases and 29 genes, 17 of which show neuronal expression, between 29.6 and 30.2 Mb on chromosome 16. In contrast, the “distal” CNVs involve the BP2–BP3 region (OMIM #613444), covering ~220 kb between 28.8 and 29 Mb (based on GRCh37/hg19) (Fig. 1). The estimated prevalence in the general population is 1 in 2000 for the typical deletion and 1 in 4100 for distal deletions. However, these figures are likely underestimated in clinical practice, partly due to limited awareness among clinicians regarding the variable expressivity and potential consequences of 16p11.2 CNVs. Notably, many parents carrying the same CNV as their affected child may show only mild or subclinical features. The majority of typical deletions are de novo (~93%), although there is evidence for dominant inheritance from an affected mother or father. Environmental and parental modifiers contributing to the wide phenotypic range remain poorly defined in the current literature.3,4
Although the 16p11.2 deletion has been extensively studied in various populations, data from Turkish cohorts remain limited. In this study, we present a single-centre retrospective analysis of 25 molecularly confirmed Turkish patients with 16p11.2 deletions. Our aim is to characterize the clinical and genetic spectrum observed in this cohort and contribute to the growing body of literature regarding genotype-phenotype correlations. Understanding these variations can ultimately guide earlier diagnosis and more tailored testing and management strategies.
Materials and Methods
Patient selection
We performed a retrospective analysis of the patients who were referred to our diagnostic centre for genetic diseases for molecular diagnosis between 2022 and 2025. All patients included in the study were referred for routine clinical indications, which included a wide variety of presenting features, such as neurological delay, speech delay, obesity, autism, dysmorphism or a combination of these symptoms. All routine clinical investigations and tests were performed in accordance with the ethical principles of the Declaration of Helsinki. Written informed consent was obtained from the patients or their legal guardians. This study was approved by the Ethics Committee of Ankara Etlik City Hospital (date: 16.07.2025, number: AEŞH-BADEK1-2025-280).
Generation and interpretation of the genetic data
All relevant genetic tests were performed in our genetic diagnosis centre using devices and kits provided by different manufacturers, following the manufacturer’s instructions, with genetic material obtained from peripheral blood cells. For chromosomal microarray analysis we used two different platforms; Infinium Global Screening Array Cyto (GSA-Cyto) chips on the Illumina iScan platform-NxClinical (v.6.0) by using Biodiscovery- Human Genome Build GRCh37 (hg19) and, CytoScan HT-CMA chips on the GeneTitan MC Fast Scan platforms Chromosome Analysis Suite (ChAS) and Reproductive Health Research Analysis Software (RhAS) using the Human Genome Build GRCh38 (hg38) reference genome. Some patients also underwent clinical exome sequencing for additional symptoms, using the Clinical Exome Solution By Sophia Genetics kit, including mitochondrial DNA sequencing, on the NovaSeq Platform (Illumina, USA) for next-generation sequencing analysis (NGS). Additional tests included methylation multiplex ligation-dependent probe amplification (MLPA) for Prader-Willi / Angelman syndromes (SALSA® MLPA® Probemix ME028 Prader-Willi / Angelman; MRC Holland, Netherlands) and trinucleotide repeat analysis for FMR1 gene repeat region. Potential causative variants that had <20X reads on NGS were confirmed with the Sanger sequencing before reporting. A lift-over process was applied for chromosome designations according to GrCh38 in tables. Patients who could not be evaluated for specific features were indicated as ‘NE: Not Evaluated’ or ‘NA: Not available’. For the interpretation of percentile-based anthropometric measurements, we used the growth reference curves standardized for Turkish children by Neyzi et al. and applied the standard body mass index (BMI) classification for individuals aged 18 years and older.5
Statistical analysis
Descriptive statistics were used to summarize the clinical and demographic characteristics of the cohort. Categorical variables are presented as frequencies and percentages. For continuous variables, such as age at diagnosis, data are expressed as median values accompanied by the interquartile range (IQR, Q1–Q3) to describe the distribution.
Results
A total of 25 patients with molecularly confirmed 16p11.2 deletions were evaluated (13 [52%] males, 12 [48%] females, and 7 [28%] familial cases). The age of the probands at diagnosis ranged from the neonatal period to 21 years, with a median age of approximately 9 years (Q1-Q3: 2.0-11.5 years). Only one proband was older than 18 years of age at the time of molecular diagnosis (P15). The majority of the confirmed cases (23, 92%) had the ‘typical’ ~600 kilobase deletion, while the distal ~220 kb BP2–BP3 deletion was detected only in P6 and her mother. P2 could not be evaluated comprehensively for many clinical features due to early-onset aspiration and subsequent severe neurological sequelae, while P7 passed away on the second postnatal day, limiting the availability of detailed phenotypic data. Autism spectrum disorder (ASD) and/or attention deficit hyperactivity disorder (ADHD) was diagnosed or strongly suspected in nearly half of the cohort (13, 52%).
Neurodevelopmental and cognitive findings
Developmental delay, mainly speech and learning problems, was the dominant feature among patients (18, 72%), with expressive language more severely affected than receptive abilities. Intellectual disability (ID) and/or learning difficulties were documented in 17 patients (68%). Most patients had mild or mild-moderate intellectual disability. Neuromotor delay and hypotonia were frequent, in terms of neuromotor development, 10 (40%) of the patients showed varying degrees of delay and, only five (20%) also had hypotonia in early infancy. Brain magnetic resonance imaging (MRI) abnormalities were observed in several individuals, including hypoplasia of the corpus callosum, idiopathic intracranial hypertension, and nonspecific gliotic changes. MRI was available for 14 cases (56%) and only five of these (35.7%) had brain abnormalities. Macrocephaly was identified in five cases (20%), three of whom (12%) overlapped with the group with brain anomalies were detected on MRI.
Epilepsy and seizure disorders
Epilepsy or recurrent seizures occurred in 13/25 patients (52%). EEG findings demonstrated generalized epileptic activity in several cases, while others showed focal discharges. Two patients had febrile seizures, and one presented with absence seizures.
Psychiatric and behavioural features
Five patients (20%) in the cohort had a diagnosis of autism spectrum disorder and eight (32%) of them had attention deficit and/or hyperactivity without symptoms or a diagnosis of autism. Ten (40%) of the patients had a history of psychiatric problems such as repetitive stereotypies, aggressive behaviour, obsessive–compulsive disorder, and tics. Psychiatric manifestations frequently co-occurred with ID and/or language impairment.
Growth and metabolic features
Overweight or obesity was observed in 10/25 patients (40%). Four (16%) of the cases had hyperphagia without obesity. Hepatic steatosis, insulin resistance, endocrine problems and abnormalities of lipid metabolism were additional metabolic findings. Conversely, a subset of patients presented with underweight or normal growth trajectories. Two of the cases had short stature.
Dysmorphic and skeletal features
Varying degrees of dysmorphic features were reported in 11 (44%) patients, albeit without a consistent pattern. Common findings included synophrys, wide or downslanting palpebral fissures, deep philtrum, and a thin upper lip. Aside from dysmorphic findings skeletal manifestations included pes equinovarus in 5 (20%) patients and scoliosis in one patient.
Other systemic findings
Ophthalmologic problems, including strabismus, optic disc changes, and papilledema were documented in several patients. Only three (12%) patients had hearing loss and cardiac examinations were normal in all patients in whom they were available. Constipation was recurrent in four (16%) cases across the cohort. Less frequent findings included hepatosplenomegaly, primary amenorrhea, and cleft palate. Ten (40%) of the probands had a history of prenatal abnormal findings or problems at birth such as polyhydramnios, prenatal macrocephaly, hyperechogenic bowel, preeclampsia, fetal distress, preterm labour, abnormal presentation, meconium aspiration.
Inheritance patterns
Parental testing revealed seven (28%) familial cases, of which five were maternally inherited and two were paternally inherited. Most of the carrier parents were either mildly affected or asymptomatic. Interestingly, some carrier parents displayed mild features (such as obesity, constipation, or subtle cognitive / behavioural issues), underscoring variable expressivity. In 14 cases (56%) parental microarray studies were not available; among the remaining cases, four (36%) were found to be de novo inheritances. Detailed characteristics of the cohort are presented in Table I, Table II, and Supplementary Table S1.
|
Age and age at diagnosis are expressed as years, unless indicated otherwise. *Patient deceased on the second day of life. CES: Clinical Exome Sequencing, DNM: de novo variation, F: Female, M: Male, MLPA: Multiplex Ligation-dependent Probe Amplification, MO: months old, N: Normal, NA: Not available, VUS: Variant of unknown significance, WES: Whole Exome Sequencing. |
|||||||
| Table I. Demographic and additional genetic information of patients. | |||||||
| Patient ID | Age | Sex | Age at diagnosis | Inheritance | Additional genetic test | Consanguinity | Additional Genetic Findings and Pathogenicity |
| P1 | 8 | M | 6 | DNM | Karyotype N FMR1 Repeat N Angelman-Prader Willi MLPA N |
- | - |
| P2 | 2 | F | 8 mo | NA | Karyotype N WES N Mitochondrial DNA N |
- | arr[GRCh37] 4p15.32p15.31 (16824676_17818885)x3 VUS |
| P3 | 12 | M | 10 | Paternal | Karyotype N | - | - |
| P3 father | 47 | M | 47 | NA | Karyotype N | - | - |
| P4 | 11 | M | 10 |
Parental Karyotypes N Microarray NA |
Karyotype N FMR1 Repeat N |
- |
arr[GRCh37] 9p23p21.3 (11374988_23957619)x3 VUS |
| P5 | 2.5 | M | 1y | Maternal |
Karyotype N CES N |
- | - |
| P5 mother | 27 | F | 25.5 | NA | - | - | - |
| P6 | 2 | F | 2y | Maternal | Karyotype N | - | - |
| P6 mother | 30 | F | 30 | NA | Karyotype N | - | - |
| P7* | 2 days | F | Post mortem | Maternal | Karyotype N | - | - |
| P7 mother | 31 | F | 29 | NA | Karyotype N | + | - |
| P8 | 17 | F | 17 | NA | Karyotype/SHOX MLPA N | - | - |
| P9 | 3 | F | 9 mo | DNM | - | - | - |
| P10 | 3 | M | 1 | DNM | Karyotype N | - | - |
| P11 | 9 | M | 8 | Maternal | Karyotype N | - | - |
| P11 mother | 41 | F | 41 | NA | Karyotype N | + | - |
| P12 | 19 mo | M | 11 mo | DNM | Karyotype N DiGeorge syndrome FISH N CES N | - | - |
| P13 | 5 | M | 4 | Maternal | Angelman-Prader Willi MLPA N Karyotype N |
- | - |
| P13 mother | 45 | F | 45 | NA | Karyotype N | + | - |
| P14 | 15 | M | 14 | NA | Karyotype N FMR1 Repeat N | - | - |
| P15 | 21 | F | 20 | NA | Karyotype N GNAS Sequencing N |
- | - |
| P16 | 9 | M | 9 | NA | Karyotype N | - | MC4R likely pathogenic variation (exome sequencing) |
| P17 | 4 | M | 3 | Paternal | Karyotype N FMR1 Repeat N |
- | - |
| P17 father |
32 | M | 32 | NA | Karyotype N | - | - |
| P18 | 6 | F | 6 | NA | CES N | - | - |
| ADHD: Attention Deficit/Hyperactivity Disorder, ASD: Autism Spectrum Disorder, MRI: Magnetic Resonance Imaging, SD: Standard Deviation. | |||
| Table II. Overview of clinical features of cases with typical 16p11.2 BP4-BP5 deletion and distal 16p11.2 BP2-BP3 deletion in our cohort. | |||
| Clinical category | Finding |
|
|
| Neurodevelopmental & cognitive | Developmental delay (mainly speech) |
|
|
| Intellectual disability / learning difficulties |
|
|
|
| Neuromotor delay |
|
|
|
| Hypotonia |
|
|
|
| Neurological | Epilepsy / recurrent seizures |
|
|
| Abnormal brain MRI findings (evaluated in n=14) |
|
|
|
| Macrocephaly |
|
|
|
| Psychiatric & behavioural | ADHD |
|
|
| ADHD with ASD |
|
|
|
| Behavioural problems (Stereotypy, aggression, obsessive compulsive disorder, tics) |
|
|
|
| Metabolic | Overweight or obesity (>2SD) |
|
|
| Hyperphagia |
|
|
|
| Hepatic steatosis |
|
|
|
| Insulin resistance / abnormal glucose metabolism |
|
|
|
| Abnormal lipid profile |
|
|
|
| Dysmorphic & skeletal | Dysmorphic facial features |
|
|
| Pes equinovarus |
|
|
|
| Cleft palate |
|
|
|
| Short stature |
|
|
|
| Pectus anomalies |
|
|
|
| Syndactyly - polydactyly |
|
|
|
| Pes valgus |
|
|
|
| Scoliosis |
|
|
|
| Other findings | Frequent infection |
|
|
| Ophthalmologic (Papilledema, abnormal optic disc) |
|
|
|
| Strabismus |
|
|
|
| Constipation |
|
|
|
| Hearing loss |
|
|
|
| Refraction problems (myopia astigmatism) |
|
|
|
| Micropenis - buried penis |
|
|
|
| Hypospadias |
|
|
|
| Prenatal & perinatal | Abnormal prenatal history / birth findings |
|
|
Additional genetic testing was performed in some cases mainly due to unexpected clinical features or initial clinical suspicion, including karyotyping, MLPA, exome sequencing, and targeted gene panels. In a few patients, variants of uncertain significance (VUS) or unrelated findings were detected (e.g., 4p15.32 duplication in P2). P16 had molecularly proven dual genetic diagnoses with heterozygous likely pathogenic MC4R variant (NM_005912: c.496G>A p.(Val166Ile)) in addition to the 16p11.2 deletion (Table I).
Discussion
This cohort of 25 individuals with 16p11.2 deletions highlights the remarkable clinical heterogeneity and multisystem involvement associated with this recurrent CNV. Consistent with existing literature, neurodevelopmental delays—particularly speech delay—emerged as hallmark features, present in the majority of cases. The prominence of expressive language deficits, often more severe than receptive language difficulties, aligns with previously published cohorts that emphasize language impairment as a key diagnostic clue. In our genetic diagnostic centre, the chromosomal microarray cohort comprises approximately 4,500 samples, and the 16p11.2 deletion was identified as the most prevalent deletion.
The recurrent 16p11.2 microdeletion encompasses approximately 29 genes, several of which have been implicated in neurodevelopment, energy homeostasis, and synaptic function, thereby contributing to the variable clinical spectrum observed in affected individuals. Among these, KCTD13 has been highlighted as a key dosage-sensitive gene influencing brain volume (micro / macrocephaly) and neurodevelopmental outcomes, especially within the autism spectrum, with both animal and human studies demonstrating its role in neurocognitive phenotypes.6 MAPK3, a critical component of the MAPK/ERK signalling pathway, is involved in neuronal differentiation, synaptic plasticity, and learning processes, and its haploinsufficiency has been associated with intellectual disability and behavioural abnormalities.7 KIF22 expression is confined to proliferating cells and peaks during mitosis, whereas ALDOA is ubiquitously expressed across cell types with stable expression throughout the cell cycle, collectively supporting the hypothesis that disrupted cortical neurogenesis may contribute to ASD in individuals with 16p11.2 CNVs.8 Additionally, DOC2A and TAOK2 are involved in synaptic vesicle trafficking and neuronal migration, respectively, and are considered contributors to the autism spectrum disorder and behavioural phenotypes observed in this deletion.9,10 Metabolic and obesity-related features frequently reported in 16p11.2 deletion carriers may be partially explained by genes such as SH2B1, located in BP2-3 region, which plays a role in leptin and insulin signalling pathways and has been linked to abnormal weight gain and the regulation of energy balance.11 While several studies have suggested that obesity is independent of the neuropsychiatric phenotypes frequently observed among CNV carriers, and no single gene or gene set within the BP4–BP5 region has been definitively linked to obesity, recent reports nevertheless hint at a potential interplay between metabolic and neurological phenotypes.12,13 The combined haploinsufficiency of these genes likely underlies the marked phenotypic variability and incomplete penetrance observed among carriers, including those with inherited deletions, as demonstrated in our cohort.
The most common symptoms in the cohort were cognitive impairments (17, 68%), speech delay observed in 18 cases (72%), seizure disorders (13, 52%) and obesity (10, 40%) (Table II). Neurodevelopmental impairments are observed in the vast majority of individuals with 16p11.2 deletions and often serve as the primary reason for initiating genetic evaluation. Most patients had either mild-to-moderate intellectual disability or significant learning difficulties. Epilepsy and abnormal EEG findings were present in more than half of the cohort. Seizure types varied, and although some were transient or febrile, others showed more complex patterns, emphasizing the need for routine neurological screening and longitudinal follow-up.
In line with the “mirror phenotype” hypothesis, macrocephaly and obesity were seen in many deletion carriers, whereas duplications, which were not analysed in the current study, are more often associated with microcephaly and low BMI.14 Almost half of the cohort was overweight or had obesity highlighting the metabolic risk associated with this deletion. Deletions in this region have been proposed in some publications as the second most common genetic cause of obesity after point mutations in the MC4R and previous studies have concluded that individuals carrying this deletion face a markedly elevated risk—estimated at 43 times higher—of developing morbid obesity, reflecting its high penetrance.15,16
Behavioural and psychiatric comorbidities were noted in half of the cohort. Isolated ADHD was only noted in eight (32%) cases while another five (20%) of the cases had a history of both ASD and ADHD. Heterozygous deletions in this region represent one of the most frequently identified genetic risk factors for autism spectrum disorder, occurring in approximately 0.5% of large ASD cohorts.17 Other psychiatric symptoms such as aggression, stereotypy, and obsessive-compulsive behaviour were also observed. These findings underscore the importance of comprehensive psychiatric evaluation and early intervention in children with 16p11.2 deletions. Neuropsychiatric atypia together with pervasive language disorders in these individuals may result in an underestimation of intelligence potential, although severe intellectual disability is reported to be rare. There is no single candidate gene for the neurodevelopmental features of proximal 16p11.2 deletions; rather, studies implicate complex, potentially synergistic interactions between several encompassed genes with some evidence for sex-specific neuroanatomical characteristics.18 The 16p11.2 deletion has thus far been reported from Türkiye in a limited number of studies, derived primarily from autism-related or autism-diagnosed cohorts.19,20 In this context, our study uniquely encompasses both a broader patient population and a more comprehensive analysis of genotype-driven phenotype correlations focused specifically on 16p11.2 deletions.
In addition to the typical ~600 kb BP4–BP5 deletions, our cohort included two individuals (P6 and her mother) with a smaller, approximately 220–269 kb deletion spanning the distal 16p11.2 BP2–BP3 region. These distal deletions are less frequently reported in the literature and are thought to have lower penetrance for neurodevelopmental disorders compared with the typical proximal deletions.21 However, our findings suggest that clinical involvement may still be significant. P6 presented with severe expressive and receptive language delay, global neuromotor delay with lack of ambulation by age 2, and moderate intellectual disability. Behavioural signs such as stereotypic movements were also observed. Interestingly, her mother, who carried the same CNV, was mostly asymptomatic aside from a high BMI (31.3) and hepatic steatosis. This familial observation further supports the notion of variable expressivity and incomplete penetrance associated with BP2–BP3 deletions. The stark phenotypic difference between the child and her carrier mother underscores the need for careful clinical evaluation and long-term follow-up even in cases in which parental carriers appear unaffected.
Dysmorphic features and congenital anomalies, while not universal, supported clinical suspicion in many patients. These findings further emphasize that the clinical diagnosis of 16p11.2 deletion cannot rely solely on syndromic appearance. Skeletal abnormalities, endocrine findings, or ophthalmologic involvement necessitate multidisciplinary care. Interestingly, nearly half of the patients had a history of abnormalities during the prenatal period or problems at birth. This may also be important for precision medicine since probands with a 16p11.2 deletion may require closer follow up during the pregnancy and birth.
One of the important associations regarding the 16p11.2 deletion is the potentially increased risk of neuroblastoma observed in affected individuals. Although studies have shown that the risk of detecting the 16p11.2 deletion is significantly higher in children with neuroblastoma compared with control groups without the disease, the fact that this lethal cancer type is observed in only a very small proportion of individuals with the 16p11.2 deletion suggests that the deletion alone is not sufficient for oncogenesis.22 Moreover, increased neuroblastoma risk has also been reported in association with other copy number variations beyond the 16p11.2 region.23-25 In our cohort, none of the molecularly confirmed individuals had been diagnosed with neuroblastoma by the time of this study.
A particularly important observation was the presence of inherited deletions in several families, where carriers were either asymptomatic or mildly affected. This reinforces the concepts of variable expressivity and reduced penetrance, which are critical for genetic counseling. In some cases, family history revealed neuropsychiatric conditions or growth abnormalities in relatives without genetic confirmation, which may suggest underdiagnosis in adult carriers (Supplementary Table S1). In the cohort, parental origin could be determined in seven cases. While no clear parental sex bias has been consistently reported in the literature, the maternal predominance in our sample may warrant further investigation in larger cohorts. In reported cases of 16p11.2 deletions, a de novo mechanism of occurrence is frequently observed. While the exact rates vary across different cohorts, in our cohort the de novo occurrence rate was found to be 36% (4/11 cases with available parental analysis). It should be noted, however, that a considerable proportion of families did not undergo segregation analysis.
With the increasing accessibility of genetic testing, multilocus genomic variants, including dual diagnosis, double diagnosis, and concurrent genetic disorders) have become more readily encountered and are now familiar topics for professionals in the field of genetics. The literature already includes various publications offering different perspectives, furthermore, we previously conducted a comprehensive study on this subject.26-28 Notably, even within this specific and technically limited cohort, a dual-diagnosis case was identified: an individual carrying both a 16p11.2 deletion and a heterozygous likely pathogenic MC4R variant (P16). We acknowledge that this finding may act as a phenotypic modifier, given that 16p11.2 deletions themselves represent one of the most penetrant genetic risks for obesity, the coexistence of these two genetic factors was interpreted as potentially additive rather than confounding. In such patients, clinical management and the concept of precision medicine gain even greater importance.
The main limitation of this study is that segregation analyses could not be performed for a substantial proportion of probands’ families. Consequently, it was not possible to provide an accurate estimate of the number of de novo versus familial cases in the cohort. In addition, for two patients, one who died in the early postnatal period (P7) and another who was affected by perinatal complications (P2), reliable correlations between phenotypic features and the deletion could not be established. Additionally, in some individuals, such as P2 and P15, the clinical presentation was more complex than typically observed in classic 16p11.2 deletion syndrome. Although further genetic investigations were conducted, no additional molecular findings were identified that could fully explain the expanded phenotype. However, due to the absence of more advanced genomic techniques such as whole genome sequencing or optical genome mapping in these cases, we refrained from attributing these atypical clinical features to the 16p11.2 deletion alone.
In summary, our cohort of 25 Turkish patients with 16p11.2 deletions highlights the characteristic yet heterogeneous nature of this CNV, with developmental and psychiatric features as the most consistent findings, alongside a higher epilepsy burden, and frequent obesity with systemic comorbidities. Comparisons with larger cohorts emphasize both shared patterns and population-specific variability. These observations underscore the importance of early recognition, multidisciplinary follow-up, and careful family-based counselling, particularly given the variable expressivity observed in carrier parents.
Ethical approval
The study was approved by Ethics Committee of Ankara Etlik City Hospital (date: 16.07.2025, number: AEŞH-BADEK1-2025-280).
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet 2009; 10: 551-564. https://doi.org/10.1038/nrg2593
- Vos N, Kleinendorst L, van der Laan L, et al. Evaluation of 100 Dutch cases with 16p11.2 deletion and duplication syndromes; from clinical manifestations towards personalized treatment options. Eur J Hum Genet 2024; 32: 1387-1401. https://doi.org/10.1038/s41431-024-01601-2
- Männik K, Mägi R, Macé A, et al. Copy number variations and cognitive phenotypes in unselected populations. JAMA 2015; 313: 2044-2054. https://doi.org/10.1001/jama.2015.4845
- Chung WK, Roberts TP, Sherr EH, Snyder LG, Spiro JE. 16p11.2 deletion syndrome. Curr Opin Genet Dev 2021; 68: 49-56. https://doi.org/10.1016/j.gde.2021.01.011
- Neyzi O, Günöz H, Furman A, et al. Weight, height, head circumference and body mass index references for Turkish children. Çocuk Sağlığı ve Hastalıkları Dergisi 2008; 51: 1-14.
- Golzio C, Willer J, Talkowski ME, et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 2012; 485: 363-367. https://doi.org/10.1038/nature11091
- Liu F, Liang C, Li Z, et al. Haplotype-specific MAPK3 expression in 16p11.2 deletion contributes to variable neurodevelopment. Brain 2023; 146: 3347-3363. https://doi.org/10.1093/brain/awad071
- Morson S, Yang Y, Price DJ, Pratt T. Expression of genes in the 16p11.2 Locus during development of the human fetal cerebral cortex. Cereb Cortex 2021; 31: 4038-4052. https://doi.org/10.1093/cercor/bhab067
- Richter M, Murtaza N, Scharrenberg R, et al. Altered TAOK2 activity causes autism-related neurodevelopmental and cognitive abnormalities through RhoA signaling. Mol Psychiatry 2019; 24: 1329-1350. https://doi.org/10.1038/s41380-018-0025-5
- Wang QW, Qin J, Chen YF, et al. 16p11.2 CNV gene Doc2α functions in neurodevelopment and social behaviors through interaction with Secretagogin. Cell Rep 2023; 42: 112691. https://doi.org/10.1016/j.celrep.2023.112691
- Ren D, Zhou Y, Morris D, Li M, Li Z, Rui L. Neuronal SH2B1 is essential for controlling energy and glucose homeostasis. J Clin Invest 2007; 117: 397-406. https://doi.org/10.1172/JCI29417
- Maillard AM, Hippolyte L, Rodriguez-Herreros B, et al. 16p11.2 Locus modulates response to satiety before the onset of obesity. Int J Obes (Lond) 2016; 40: 870-876. https://doi.org/10.1038/ijo.2015.247
- Tomasello DL, Kim JL, Khodour Y, et al. 16pdel lipid changes in iPSC-derived neurons and function of FAM57B in lipid metabolism and synaptogenesis. iScience 2021; 25: 103551. https://doi.org/10.1016/j.isci.2021.103551
- Jacquemont S, Reymond A, Zufferey F, et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature 2011; 478: 97-102. https://doi.org/10.1038/nature10406
- Walters RG, Jacquemont S, Valsesia A, et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature 2010; 463: 671-675. https://doi.org/10.1038/nature08727
- Atli EI, Yalcintepe S, Atli E, Demir S, Mail C, Gurkan H. Clinical implications of chromosome 16 copy number variation. Mol Syndromol 2022; 13: 184-192. https://doi.org/10.1159/000517762
- Walsh KM, Bracken MB. Copy number variation in the dosage-sensitive 16p11.2 interval accounts for only a small proportion of autism incidence: a systematic review and meta-analysis. Genet Med 2011; 13: 377-384. https://doi.org/10.1097/GIM.0b013e3182076c0c
- Kretz PF, Wagner C, Mikhaleva A, et al. Dissecting the autism-associated 16p11.2 locus identifies multiple drivers in neuroanatomical phenotypes and unveils a male-specific role for the major vault protein. Genome Biol 2023; 24: 261. https://doi.org/10.1186/s13059-023-03092-8
- Gümüşlü KE, Savli H, Sünnetçi D, et al. A CGH array study in nonsyndromic (primary) autism patients: deletions on 16p13.11, 16p11.2, 1q21.1, 2q21.1q21.2, and 8p23.1. Turk J Med Sci 2015; 45: 313-319.
- Özaslan A, Kayhan G, İşeri E, Ergün MA, Güney E, Perçin FE. Identification of copy number variants in children and adolescents with autism spectrum disorder: a study from Turkey. Mol Biol Rep 2021; 48: 7371-7378. https://doi.org/10.1007/s11033-021-06745-8
- Kim SH, Green-Snyder L, Lord C, et al. Language characterization in 16p11.2 deletion and duplication syndromes. Am J Med Genet B Neuropsychiatr Genet 2020; 183: 380-391. https://doi.org/10.1002/ajmg.b.32809
- Egolf LE, Vaksman Z, Lopez G, et al. Germline 16p11.2 microdeletion predisposes to neuroblastoma. Am J Hum Genet 2019; 105: 658-668. https://doi.org/10.1016/j.ajhg.2019.07.020
- Nagano H, Kano Y, Kobuchi S, Kajitani T. A case of partial 2p trisomy with neuroblastoma. Jinrui Idengaku Zasshi 1980; 25: 39-45. https://doi.org/10.1007/BF01876544
- Isidor B, Le Cunff M, Boceno M, et al. Complex constitutional subtelomeric 1p36.3 deletion/duplication in a mentally retarded child with neonatal neuroblastoma. Eur J Med Genet 2008; 51: 679-684. https://doi.org/10.1016/j.ejmg.2008.06.004
- Koiffmann CP, Gonzalez CH, Vianna-Morgante AM, Kim CA, Odone-Filho V, Wajntal A. Neuroblastoma in a boy with MCA/MR syndrome, deletion 11q, and duplication 12q. Am J Med Genet 1995; 58: 46-49. https://doi.org/10.1002/ajmg.1320580110
- Kablan A, Sezer A, Bakır A, et al. Multilocus disease-causing genomic variations for genetic disorders: single tertiary centre experience from Türkiye. Clin Genet 2025; 108: 240-258. https://doi.org/10.1111/cge.14740
- Posey JE, Harel T, Liu P, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med 2017; 376: 21-31. https://doi.org/10.1056/NEJMoa1516767
- Narayanan DL, Udyawar D, Kaur P, et al. Multilocus disease-causing genomic variations for Mendelian disorders: role of systematic phenotyping and implications on genetic counselling. Eur J Hum Genet 2021; 29: 1774-1780. https://doi.org/10.1038/s41431-021-00933-7
Copyright and license
Copyright © 2026 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









