
Abstract
Background. Primary hyperparathyroidism (PHPT) is a rare endocrine disorder in childhood, most commonly associated with a single parathyroid adenoma. Compared to adults, pediatric cases often present with more pronounced clinical manifestations and may lead to severe skeletal complications. This report presents a symptomatic case of PHPT complicated by extensive skeletal involvement, brown tumors, and postoperative hungry bone syndrome (HBS).
Case Presentation. A 16-year-old female was admitted with progressive leg pain and weight loss. Laboratory evaluation revealed marked hypercalcemia, severely elevated parathyroid hormone, hypophosphatemia, and vitamin D deficiency. Imaging findings were consistent with a parathyroid adenoma; however, the initial surgical attempt failed to localize the adenoma. Subsequent advanced imaging with four-dimensional computed tomography (4D-CT) and interventional radiology-guided localization enabled successful resection. Postoperatively, the patient developed profound and prolonged hypocalcemia with concomitant hypophosphatemia and hypomagnesemia, consistent with HBS, requiring intensive intravenous and oral calcium, calcitriol, phosphate, and magnesium replacement. Radiological and histopathological evaluations demonstrated diffuse skeletal involvement with multiple brown tumors.
Conclusions. This case highlights that although rare, PHPT should be considered in the differential diagnosis of children presenting with refractory bone pain and hypercalcemia. Accurate preoperative localization of parathyroid adenomas requires advanced imaging techniques and a multidisciplinary approach. Furthermore, in patients with markedly elevated parathyroid hormone and alkaline phosphatase levels, vitamin D deficiency, and long-standing skeletal involvement, the risk of developing HBS should be anticipated and management strategies tailored accordingly.
Keywords: primary hyperparathyroidism, parathyroid adenoma, hypercalcemia, brown tumors, hungry bone syndrome
Introduction
Primary hyperparathyroidism (PHPT) is a condition characterized by hypercalcemia due to excessive secretion of parathyroid hormone (PTH) from the parathyroid glands, accompanied by inappropriately normal or elevated serum PTH levels. While PHPT is more common in adults, it is rare in childhood and adolescence, with an incidence of 2–5 cases per 100,000 individuals.1,2 The most common cause of PHPT in childhood and adolescence is a single parathyroid adenoma, accounting for 70–80% of cases. Multiglandular adenomas or multiple adenomas, are less common, accounting for approximately 15–20% of cases. Adenomas are usually sporadic, and some cases may develop with multiple endocrine neoplasia (MEN) type 1 and 2A syndromes or familial isolated hyperparathyroidism.3
Symptoms of hyperparathyroidism include constipation, bone pain, fatigue, and depression. Severe hypercalcemia can cause shortening of the QT interval (the time from the Q wave of the QRS complex to the end of the T wave) on the ECG, ventricular tachycardia, and even fatal arrhythmias such as ventricular fibrillation. Hypercalcemic patients with hyperparathyroidism may experience nephrolithiasis, osteopenia, osteoporosis, bone fractures, pancreatitis, peptic ulcer disease, and hypertension. Symptomatic disease is more common in children with primary hyperparathyroidism than in adults (79%–90%), and 44% of organ involvement (e.g., nephrolithiasis, nephrocalcinosis, and bone involvement) can be definitively treated with surgery. Pediatric case reports demonstrate a wide range of nonspecific features.4,5
Brown tumors are rare focal giant cell lesions that arise from hypercalcemia resulting from excessive PTH secretion from the parathyroid glands. These lesions are caused by the direct effect of PTH on bone tissue in patients with hyperparathyroidism. In normal physiology, the parathyroid glands continuously monitor serum calcium levels and increase PTH secretion in response to hypocalcemia. Elevated PTH levels increase serum calcium levels by increasing calcium reabsorption in the kidneys and bone resorption. However, in pathological conditions, this mechanism operates uncontrolled, leading to aggressive osteoclastic activity in bone tissue and the development of localized lytic lesions. Histologically, these lesions exhibit a characteristic “brown” color due to vascularity, hemorrhage, and hemosiderin deposition. Although nonneoplastic, they can be locally destructive.6,7
Hungry bone syndrome (HBS) is a clinical condition characterized by pronounced and prolonged hypocalcemia that develops due to a sudden decrease in PTH levels in patients who underwent parathyroidectomy for PHPT and had high preoperative bone turnover. This condition is frequently accompanied by hypophosphatemia and hypomagnesemia. Increased calcium transfer into bone tissue as a result of sudden PTH suppression is proposed as the primary pathophysiological mechanism, and this is associated with a decrease in bone remodeling processes. Although HBS is rare after parathyroidectomy, it is a complication that can lead to serious clinical consequences.8,9 Although current literature data is limited, it is estimated that approximately 13% of patients undergoing parathyroidectomy may develop HBS. Risk factors that may influence the development of HBS include young age at the time of surgery, elevated PTH and alkaline phosphatase (ALP) levels, low serum albumin and magnesium levels, and skeletal pathologies such as subperiosteal erosions, lytic bone lesions, and brown tumors.10
This report aims to present an exceptionally rare and severe case of pediatric primary hyperparathyroidism characterized by extensive skeletal destruction, multiple brown tumors, and postoperative hungry bone syndrome, and to underscore the critical importance of timely diagnosis, precise preoperative localization, and proactive postoperative management in this vulnerable age group.
Case Presentation
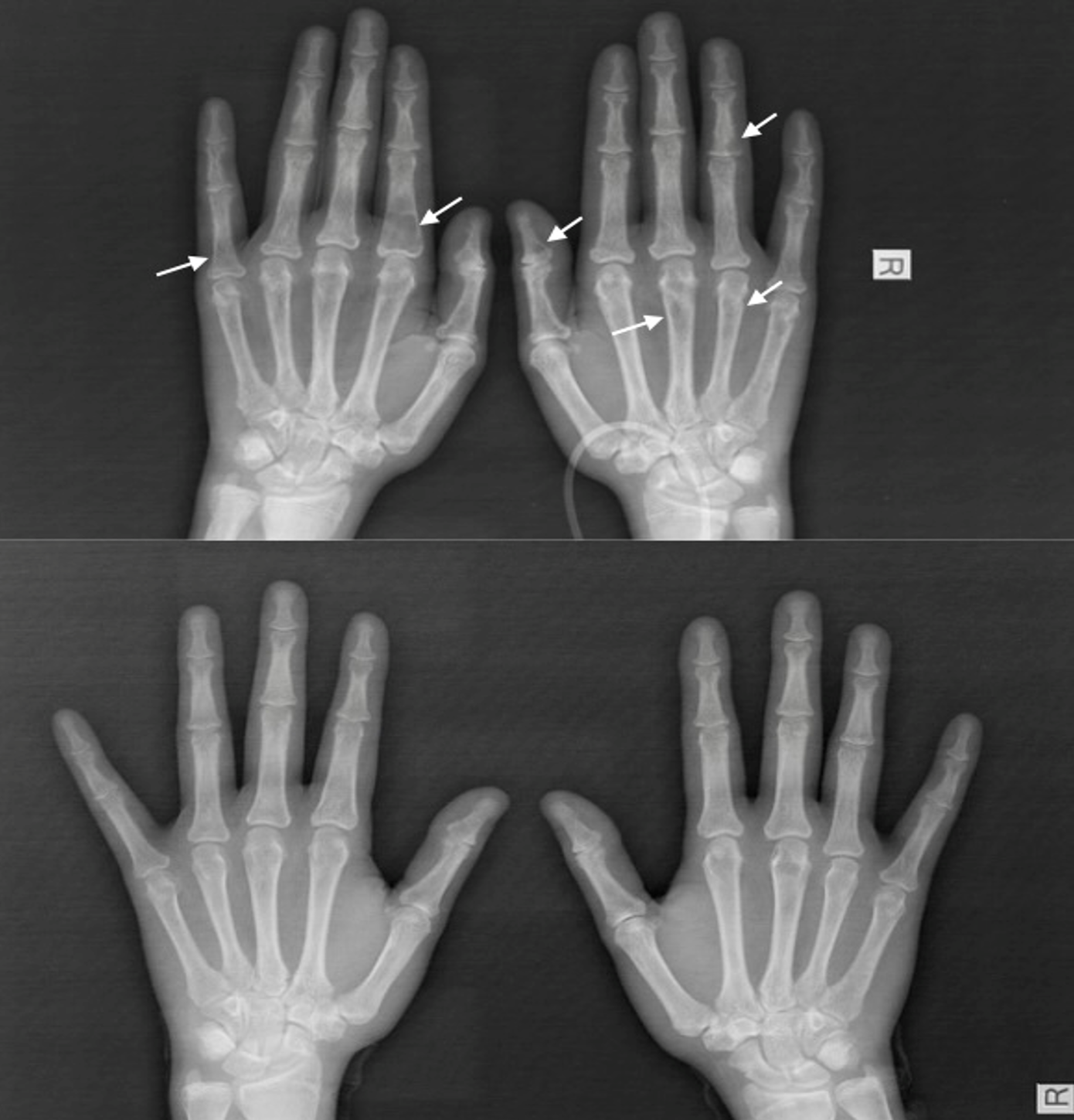
We present the case of a 16-year-old female who had been initially admitted to another health center with complaints of progressively worsening diffuse leg pain and weight loss over the past two months. Initial evaluation revealed a serum calcium level of 12.9 mg/dL (8.4-10.2), an ALP level of 983 U/L (50-117), and a PTH level of 1127 pg/mL (15-65). Despite intravenous (IV) hydration and furosemide therapy for hypercalcemia, serum calcium levels persisted, leading to the plan to begin pamidronate therapy. An allergic reaction developed during the pamidronate infusion, and treatment was discontinued. Neck ultrasonography (USG) revealed a hypoechoic, 20x30 mm solid lesion adjacent to the inferior left thyroid pole, demonstrating vascularization, and was diagnosed as a parathyroid adenoma. A lower extremity magnetic resonance imaging (MRI) was performed due to widespread persistent lower extremity bone pain, which revealed widespread heterogeneous signal intensities in the proximal and mid-portions of both tibias, focal sclerotic areas in the left epiphyseal-metaphyseal region, and an infiltrative lesion (Fig. 1). The preliminary diagnosis was Langerhans cell histiocytosis or brown tumor. The patient was referred to our center for further evaluation by the pediatric endocrinology, oncology, and surgery departments. The patient’s history did not include any prenatal, natal, or postnatal features. She had no known chronic disease or regular medication use. There was no family history of consanguinity between the mother and father. It was learned that the mother was under treatment for hyperthyroidism, and the father and a sister were under treatment for systemic sclerosis. On physical examination, her body weight was 43 kg (−2.17 SDS), height was 155 cm (−1.19 SDS), and body mass index was 17.9 kg/m2 (−1.61 SDS). Pubertal development was consistent with Tanner stage 5. Systemic examination findings were normal. Laboratory findings revealed a serum calcium level of 13 mg/dL (8.4-10.2), serum phosphate was 2.2 mg/dL (2.8-4.8), serum magnesium was 2.01 mg/dL (1.7-2.2), serum ALP was 938 U/L (50-117), urine calcium/creatinine ratio was 0.7 mg/mg, serum intact PTH level was 1127 pg/mL (15-65), and there was severe vitamin D deficiency (serum 25-OH-D3 level was 4.77 ng/mL). Diffuse radiolucent areas on whole-body plain radiographs were interpreted as brown tumors (Fig. 2). On the 16th day of hospitalization, while the patient was undergoing preoperative preparation for hyperparathyroidism due to a parathyroid adenoma, a spontaneous subtrochanteric femoral fracture occurred. Dual-energy X-ray absorptiometry (DXA) revealed a lumbar spine (L1–L4) bone mineral density Z-score of −3.5. Based on the presence of a femoral fracture and markedly reduced bone mineral density, the patient was diagnosed with secondary pediatric osteoporosis. Calcitonin levels (screened for MEN 2A), urinary catecholamine and metabolite levels (screened for pheochromocytoma / MEN 2A), and the anterior pituitary hormone profile (screened for MEN 1) were all within normal limits. Parathyroid scintigraphy revealed activity uptake in the lower pole of the left thyroid lobe consistent with a parathyroid adenoma. The patient, diagnosed with PHPT and symptomatic hypercalcemia due to a parathyroid adenoma, was operated on by a pediatric surgeon based on the current ultrasound and scintigraphy imaging findings.
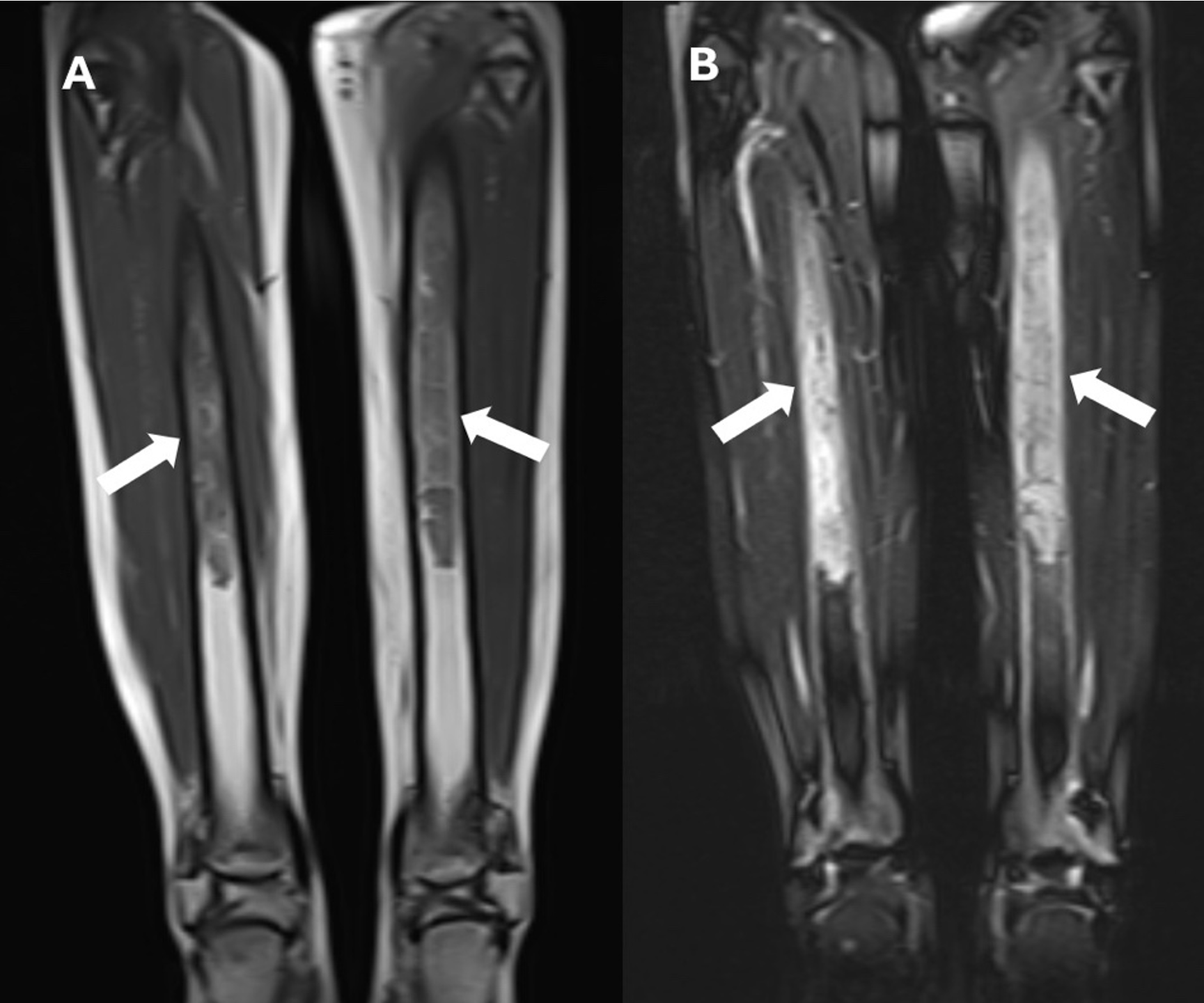
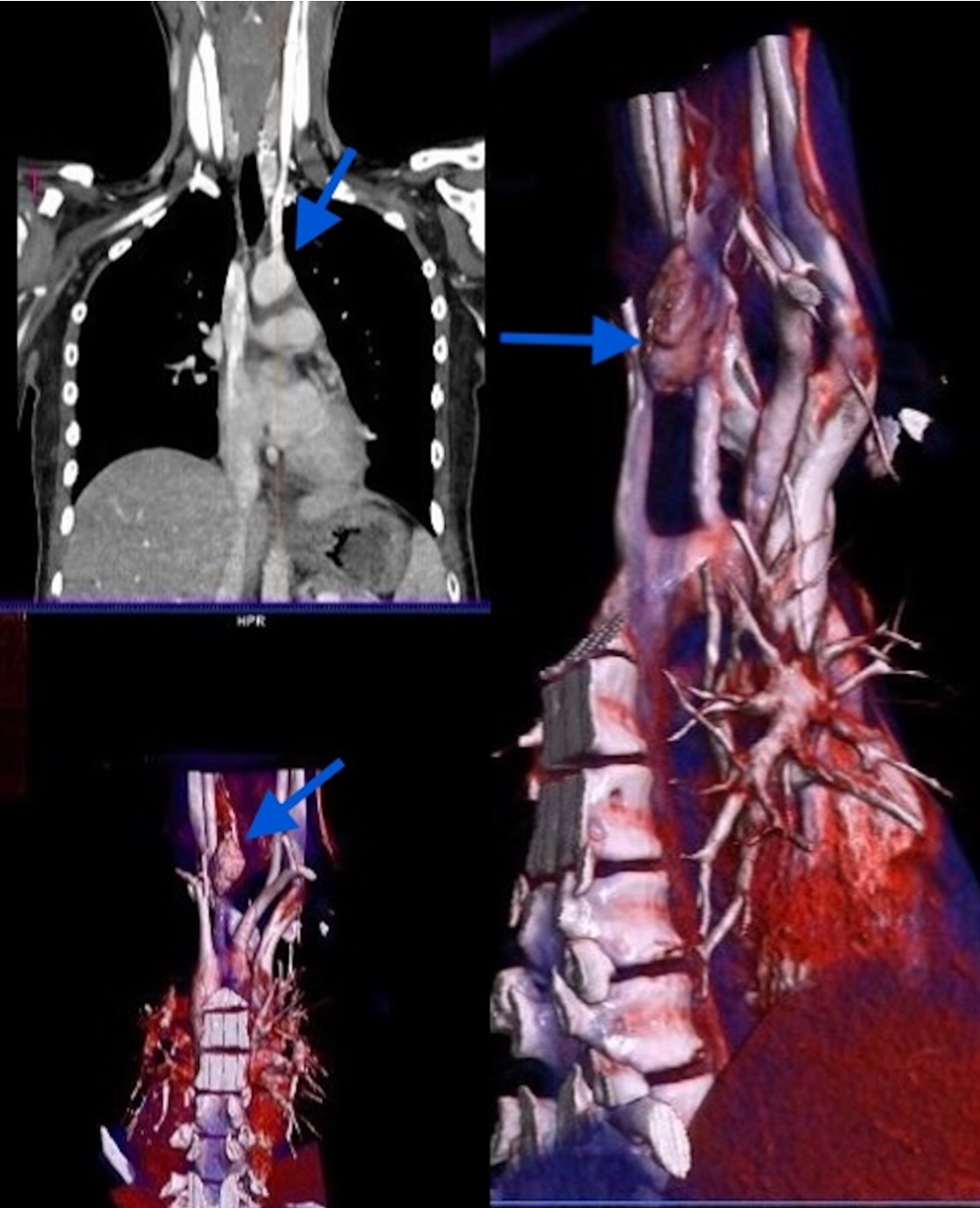
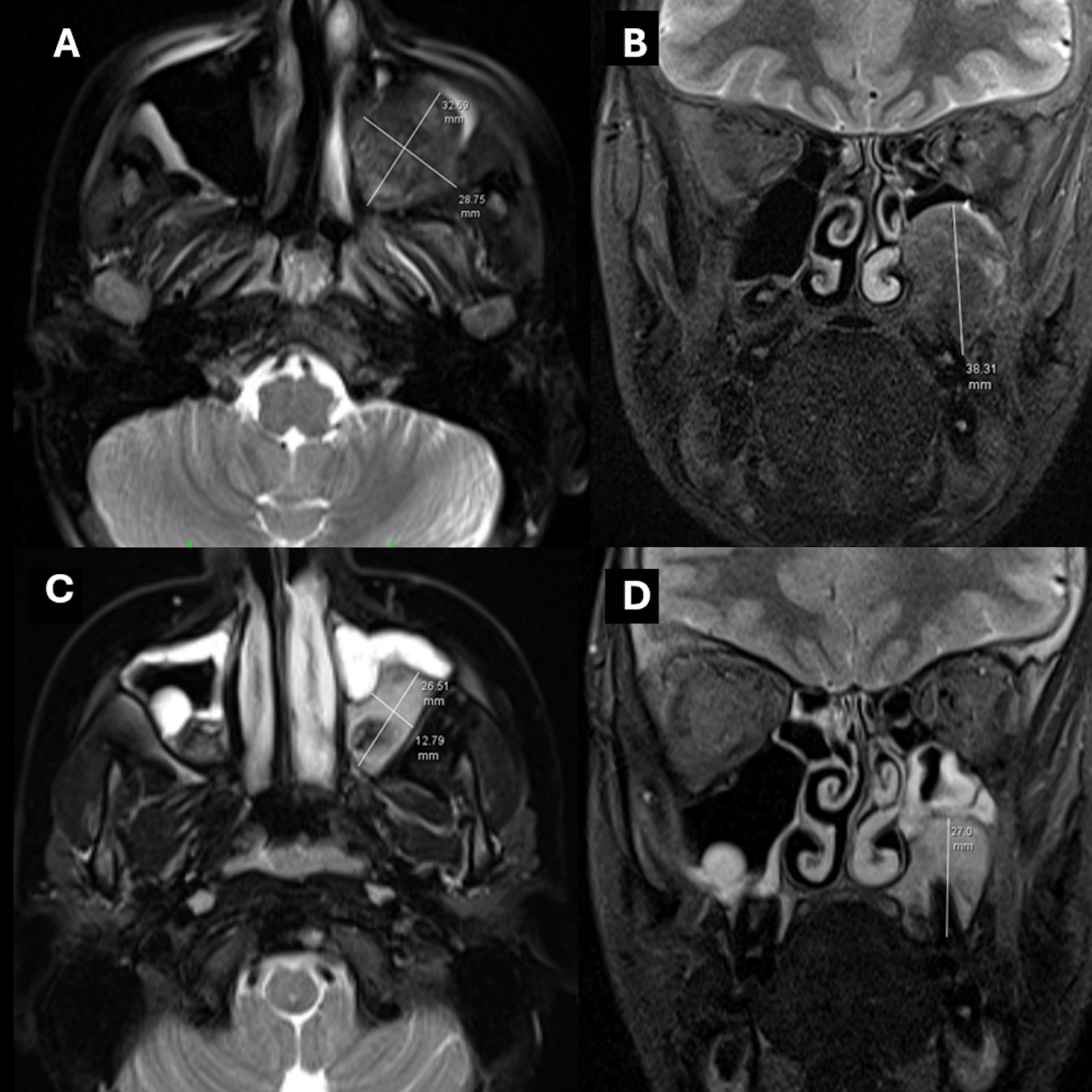
The patient with persistent postoperative hypercalcemia and elevated serum PTH levels underwent dynamic computed tomography (4D-CT) of the neck and thorax. Imaging demonstrated a mass lesion situated posterolateral to the trachea and mediolateral to the left common carotid artery (Fig. 3). Diffuse sclerotic bone changes were noted in the vertebral column, ribs, humerus, clavicle, and scapula. Furthermore, a heterogeneous mass measuring 30 × 35 × 30 mm, containing sclerotic components, was identified along the posteromedial wall of the left maxillary sinus (Fig. 4). Histopathological examination of a biopsy specimen obtained from the maxillary lesion revealed findings consistent with a brown tumor.
Before the planned second operation, interventional radiology marked the adenoma location. The surgical team accessed the lesion with a guidewire and successfully excised the adenoma. In the postoperative period, the patient developed marked hypocalcemia accompanied by low parathyroid hormone levels (serum calcium: 5.2 mg/dL; PTH: 14.8 pg/mL). The clinical presentation was considered consistent with transient postoperative hypoparathyroidism and hungry bone syndrome. Accordingly, treatment was intensified with intravenous calcium gluconate (2.4 g/day), oral calcium carbonate (6 g/day), and calcitriol at a dose of 3 µg/day. During this period, hypophosphatemia and hypomagnesemia accompanying hypocalcemia were also detected; therefore, oral phosphate solution and magnesium supplementation were added to the treatment regimen. Stabilization of serum calcium, phosphorus, and magnesium levels was achieved by the third postoperative week. Subsequently, therapy was transitioned to an oral regimen consisting of calcium carbonate 3 g/day, calcitriol 2 µg/day, and magnesium 365 mg administered three times daily, and the patient was discharged on this treatment.
During the follow-up period, no clinical symptoms related to hypocalcemia, bone pain, or new fractures were observed. Treatment doses were gradually tapered based on serial serum calcium and magnesium levels, as well as the urinary calcium-to-creatinine ratio. At the first postoperative month, biochemical evaluation revealed normalization of serum calcium and parathyroid hormone levels, and the transient hypoparathyroid state was considered resolved. Given the persistently low bone mineral density, calcium, magnesium, and calcitriol supplementation were continued. With stepwise dose reduction, all treatments except 25-hydroxyvitamin D were discontinued by the eighth postoperative month. One month after cessation of therapy, biochemical assessment demonstrated a serum calcium of 9.6 mg/dL, phosphorus of 3.3 mg/dL, magnesium of 1.9 mg/dL, parathyroid hormone of 24 pg/mL, and 25-hydroxyvitamin D of 28 ng/mL. Consequently, maintenance therapy was continued with 25-hydroxyvitamin D prophylaxis at a daily dose of 600 IU.
Physical therapy was initiated to support the skeletal system in the patient, who was considered osteoporotic based on bone mineral density assessment. At the first-year postoperative follow-up, bone mineral densitometry revealed an age-adjusted lumbar spine (L1–L4) Z-score of −0.6 SDS. The patient was also followed by the orthopedic team, and removal of the femoral implant placed intraoperatively for the subtrochanteric femoral fracture was planned at three months.
Informed consent was obtained from the patient and her parents for this publication.
Discussion
This case demonstrates a symptomatic and complicated presentation of PHPT, a rare condition in childhood. PHPT is an endocrine disorder relatively rare in children compared to adults, with a reported incidence of 2–5 per 100,000. PHPT generally presents with distinct symptoms at diagnosis in the pediatric age group and can lead to systemic complications. As in the case presented here, musculoskeletal pain, anorexia, and weight loss due to hypercalcemia are common presentations in children. Our patient, who began with complaints of widespread leg pain and weight loss, was diagnosed with hyperparathyroidism due to severe hypercalcemia detected during biochemical evaluation, along with markedly elevated serum PTH levels. One of the key diagnostic factors in our patient was the coexistence of elevated PTH, low phosphorus levels, high ALP, and vitamin D deficiency. This biochemical profile helps exclude benign differentials, particularly familial hypocalciuric hypercalcemia (FHH). Furthermore, the patient’s urine calcium/creatinine ratio, as high as 0.7, also excludes FHH.11 The absence of pathological findings in genetic and hormonal screening for MEN syndromes, along with the ultrasound findings, supports the diagnosis of sporadic adenoma-induced PHPT.
The literature has reported a higher rate of symptomatic presentation in childhood PHPT cases compared to adults, with musculoskeletal findings being particularly prominent.4,11 The symptoms and distinct radiological findings of bone involvement in our patient demonstrate the destructive effects of long-term PTH exposure on the skeletal system.
MRI and CT revealed widespread heterogeneous signal changes and sclerotic areas in both tibias, the vertebral columns, ribs, humerus, and maxillary sinuses, consistent with advanced bone involvement and a mass in the maxillary sinus, suggesting a diagnosis of brown tumor. These lesions are non-neoplastic giant cell foci that develop secondary to hyperparathyroidism and exhibit pronounced osteoclastic activity. In cases of brown tumor reported by Guedes et al., the lesions, which presented with lytic and destructive appearances in bone, were histopathologically reported to be giant cell structures with hemosiderin-laden, vascular stroma.6 In our case, radiological findings, a bone mineral density z-score of -3.5, and widespread skeletal involvement support the diagnosis of brown tumor. Although such lesions are histologically benign, they can be locally aggressive and cause severe structural deformities if diagnosis is delayed. In the differential diagnosis of brown tumor, it is important to distinguish them from giant cell bone tumors and infiltrative bone diseases such as Langerhans cell histiocytosis; however, the most critical differentiating factors are the presence of concomitant marked PTH elevation and hypercalcemia.6,7 The literature has reported that brown tumors are more common in cases of long-standing hyperparathyroidism, in postmenopausal women, and after the age of 50, while they are extremely rare before puberty.6 In this context, the development of diffuse brown tumor that we described in our 16-year-old female patient is one of the rare conditions described in the literature; this can be explained by the fact that the disease progresses with advanced bone involvement, meaning that it remains undiagnosed for a long time. Bone lesions associated with brown tumors typically show regression following parathyroidectomy in the majority of cases. Effective and appropriate treatment of hyperparathyroidism leads to suppression of osteoclastic activity and is accompanied by the emergence of new bone formation.6 Consistent with the literature, follow-up brain magnetic resonance imaging performed at the eighth postoperative month in our patient demonstrated a 60% reduction in the maxillary mass lesion (Fig. 4). Furthermore, follow-up plain radiographs obtained at the twelfth month revealed a decrease in sclerotic changes in the bones, with complete resolution observed in some lesions (Fig. 2).
Another notable aspect of our case is the difficulty in localizing the adenoma and the failure of the initial surgical attempt. Preoperative accurate localization of a parathyroid adenoma is crucial, especially when minimally invasive parathyroidectomy is planned. Cervical USG and 99mTc-sestamibi parathyroid scintigraphy are traditionally the first-line imaging methods. In adult series, both methods have a sensitivity of approximately 70–85% in cases of PHPT with a single adenoma, and diagnostic accuracy increases when used together.12,13 Imaging methods can be inadequate in pediatric patients due to the small size of the glands, potential ectopic location, and unclear boundaries with the thyroid tissue. Ultrasound and Tc-99m sestamibi scintigraphy, in particular, can lead to false-negative results even in experienced hands. In such cases, advanced imaging techniques such as 4D-CT or intraoperative localization with accurate marking are critical for successful surgery.13 The adenoma was not detected in our patient’s first operation. Subsequently, 4D-CT localized the adenoma, and interventional radiology-guided localization allowed a successful resection in the second attempt. This case highlights the importance of a multidisciplinary approach and, when necessary, the use of advanced imaging techniques in PHPT, a rare condition in childhood.
The severe hypocalcemia that developed in our patient following adenoma excision was found to be consistent with hungry bone syndrome. This syndrome is a clinically significant condition characterized by increased bone resorption and turnover due to long-term PTH administration due to hyperparathyroidism. This syndrome results from the rapid absorption of calcium from the circulation by the bones following surgical excision of the adenoma. The literature reports that the incidence of HBS ranges from 25% to 90% in patients with skeletal involvement, while this rate is significantly lower at 0% to 6% in those without skeletal involvement.8,9 Particularly in the preoperative period, severe vitamin D deficiency, the presence of a widespread brown tumor, and high ALP levels placed our patient at high risk for HBS. Despite active vitamin D therapy and high-dose oral and intravenous calcium replacement therapy for hypocalcemia, which began early after surgery, hypocalcemia persisted, accompanied by concurrent hypophosphatemia and hypomagnesemia. Because magnesium deficiency suppresses PTH secretion and weakens the calcium response, ensuring appropriate doses of magnesium replacement is critical for treatment. In our patient, stabilization of serum calcium levels was achieved after approximately 10 days of intensive intravenous and oral calcium therapy. While the duration of HBS varies from patient to patient, some cases may require weeks of intravenous supplemental therapy. Therefore, risk factors for the development of HBS should be identified preoperatively, and treatable risk factors, such as vitamin D deficiency, should be corrected before surgery. Due to osteoporosis secondary to long-term hypercalcemia, our patient received physical therapy support after discharge. Due to significant osteoporosis, bisphosphonate therapy was planned to support skeletal reconstruction. Children with parathyroid adenomas who carry this level of metabolic burden after surgery require follow-up by a multidisciplinary team consisting of pediatric endocrinology, surgery, interventional radiology, and physical therapy.
Conclusion
The patient we present is a remarkably rare case, demonstrating both the diagnostic challenges and systemic complications of PHPT in the pediatric age group. Although rarely seen in children presenting with widespread bone pain, PHPT should be considered in the differential diagnosis. In patients with parathyroid adenoma, accurate preoperative localization of the adenoma, along with assessment of risk factors for complications such as hungry bone syndrome—including age, elevated PTH and ALP levels, and low vitamin D status—and appropriate postoperative management of hypoparathyroidism and bone reconstruction directly influence treatment success and the recovery process.
Ethical approval
Informed consent was obtained from the patient and her parents for this publication.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Dutta A, Pal R, Jain N, et al. Pediatric parathyroid carcinoma: a case report and review of the literature. J Endocr Soc 2019; 3: 2224-2235. https://doi.org/10.1210/js.2019-00081
- Bilezikian JP, Bandeira L, Khan A, Cusano NE. Hyperparathyroidism. Lancet 2018; 391: 168-178. https://doi.org/10.1016/S0140-6736(17)31430-7
- Stack BC Jr, Singer MC, Shonka DC Jr, editors. Medical and surgical treatment of parathyroid diseases: an evidence-based approach. Cham: Springer Nature; 2025. https://doi.org/10.1007/978-3-031-81813-4
- Roizen J, Levine MA. Primary hyperparathyroidism in children and adolescents. J Chin Med Assoc 2012; 75: 425-434. https://doi.org/10.1016/j.jcma.2012.06.012
- Jamshidi R, Egan JC. Pediatric parathyroid disease. Semin Pediatr Surg 2020; 29: 150923. https://doi.org/10.1016/j.sempedsurg.2020.150923
- Guedes A, Becker RG, Nakagawa SA, Guedes AAL. Update on brown tumor of hyperparathyroidism. Rev Assoc Med Bras (1992) 2024; 70: e2024S132. https://doi.org/10.1590/1806-9282.2024S132
- Majumdar S, Uppala D, Kotina S, Alekhya B. Brown tumor of hyperparathyroidism with multiple lesions. J Oral Maxillofac Pathol 2022; 26: S111-S115. https://doi.org/10.4103/jomfp.jomfp_409_20
- De Armas-Conde M, Camarasa-Pérez Á, García-Martínez R, Hueso-Mor A, Caballero-Rodríguez E, Jordán-Balanzá JC. Hungry bone syndrome following thyroid surgery. J Surg Case Rep 2024; 2024: rjae031. https://doi.org/10.1093/jscr/rjae031
- Escudero MO, Kuspiel MF, Eymann AM, De la Iglesia Niveyro PX, Alonso G. Atypical parathyroid tumor: a rare cause of primary hyperparathyroidism in an adolescent. Arch Argent Pediatr 2025; 123: e202410589. https://doi.org/10.5546/aap.2024-10589.eng
- Tavakoli F, Yaghoubi F, Dalil D, Rezaei M. Multiple fractures due to hungry bone syndrome following parathyroidectomy: a clinical case report and review of literature. Clin Diabetes Endocrinol 2024; 10: 25. https://doi.org/10.1186/s40842-024-00183-8
- Ray GJ, Liles JS, Lim WY. A case of primary hyperparathyroidism secondary to parathyroid adenoma in a pediatric patient. AACE Clin Case Rep 2024; 10: 97-100. https://doi.org/10.1016/j.aace.2024.02.008
- Kızılcan Çetin S, Şıklar Z, Aycan Z, et al. Clinical profile of parathyroid adenoma in children and adolescents: a single-center experience. Turk Arch Pediatr 2023; 58: 56-61. https://doi.org/10.5152/TurkArchPediatr.2022.22180
- Rampp RD, Mancilla EE, Adzick NS, et al. Single gland, ectopic location: adenomas are common causes of primary hyperparathyroidism in children and adolescents. World J Surg 2020; 44: 1518-1525. https://doi.org/10.1007/s00268-019-05362-8
Copyright and license
Copyright © 2026 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.









